
More Information
Submitted: 28 April 2020 | Approved: 20 May 2020 | Published: 21 May 2020
How to cite this article: D’Souza SM, Cundra LB, Yoo BS, Parekh PJ, Johnson DA. Microbiome and Gastroesophageal Disease: Pathogenesis and Implications for Therapy. Ann Clin Gastroenterol Hepatol. 2020; 4: 020-033.
DOI: 10.29328/journal.acgh.1001018
ORCiD: orcid.org/0000-0002-8737-0711
Copyright License: © 2020 D’Souza SM, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Microbiome; GERD; Probiotics; Prebiotics; Bacteriocins; Dysbiosis; Barrett’s esophagus; Esophageal cancer; Esophagitis; Eosinophilic esophagitis

Microbiome and Gastroesophageal Disease: Pathogenesis and Implications for Therapy
Steve M D’Souza1, Lindsey B Cundra1, Byung Soo Yoo1, Parth J Parekh2 and David A Johnson2*
1Department of Internal Medicine, Eastern Virginia Medical School, Norfolk VA, USA
2Department of Internal Medicine, Gastroenterology Division, Eastern Virginia Medical School, Norfolk VA, USA
*Address for Correspondence: David A Johnson, MD, MACG, FASGE,MACP, Professor of Medicine, Chief of Gastroenterology, Eastern VA Medical School, Norfolk VA, USA, Tel: 7574660165; Email: [email protected]
There is growing evidence that gastroesophageal disease is influenced by the esophageal microbiome, and that commensal bacteria of the oropharynx, stomach, and colon are thought to have a role in modulatiing pathogenesis. These emerging hypotheses are based on observed changes in the composition of the esophageal flora, notably, repeated observations: 1. There is an abundance of gram-positive bBacteria in the healthy esophagus. are more gram positive prevalent 2. The esophageal bacterial population becomes increasingly gram negative with disease progression. Associated with this shift to a more gram negative prevalence is an increase in the potential for the presence of antigenic lipopolysaccharide (LPS). The immunoreactivity of LPS endotoxin thought to promote susceptibility to inflammation and disease.
The pathogenesis of the more common diseases of the esophagus e.g. gastroesophageal reflux disease (GERD), esophageal dysmotility (achalasia), eosinophilic esophagitis (EoE), Barrett’s esophagus (BE), and esophageal cancer, are well-established. Emerging data suggest however, that these are all characterized by an immune-mediated inflammatory cascade, propogated by a dysbiotic state. Thereby, the ability of the healthy “normative state” to protect against foreign bacteria is compromised. This dysbiosis thereby can create adverse inflammatory or immunoregulatory responses with progression of disease.
In the normal healthy state, the esophageal microbiome is constituted in-part, by a multitude of gram positive bacteria, many of which produce antibacterial peptides called bacteriocins. Bacteriocins are selective and used to maintain population integrity by killing off foreign bacteria. When the “normative biome” is interrupted (e.g. antibiotics, medications, diet, environmental factors), the constitutional changes may allow a more hospitable imbalance favoring the proliferation of opportunistic pathogens. Therefore it seems rational that defining, perhaps that defining, perhaps cultivating, a protective bacterial community that could help prevent or mitigate inflammatory diseases of the esophagus. Furthermore, in conjunction with evidence demonstrating that some bacteriocins are cytotoxic or antiproliferative toward cancer cell lines, further exploration might provide a rich source of effective peptide-based drug targets.
Therapeutic options targeting the microbiome, including prebiotics, probiotics, antibiotics and bacteriocins, have been studied, albeit the attributable effects on the esophagus for the most part, have been unrecognized by clinicians. This review focuses on the current knowledge of the involvement of the microbiome in esophageal diseases (most notably GERD/Barrett’s esophagus/esophageal cancer) and identifies emerging new concepts for treatment.
Gastroesophageal disease is a major source of health and economic cost worldwide, and, in 2015, annual expenditures for esophageal disorders were estimated at $18.1B annually in the United States [1]. Gastroesophageal reflux disease (GERD) is the most common esophageal pathology, with 31% of the United States population reporting heartburn or reflux symptoms within the past week [1,2]. Barrett’s esophagus (BE), a premalignant complication of GERD, has an estimated prevalence of 5.6% within the United States [3]. Esophageal cancer also poses a significant burden of disease, with > 18,000 new diagnoses and > 16,000 deaths estimated in 2020 [4]. Changes in understanding of the pathogenesis of the esophageal disease have contributed to the development of new possible therapeutic options.
There has been a recent meteoric rise in the literature demonstrating the significance of the gut microbiome and dysbiosis (defined as microbial imbalance or maladaptation), in the pathogenesis of gastrointestinal (GI)disease [5]. This article aims to review the current literature for microbiome-related pathogenesis of gastroesophageal disease and to discuss disease-mitigation strategies and areas for future research.
“Normal” gastroesophageal microflora
The esophageal microbiome is shaped by the oral cavity, oropharynx, and stomach due to migration of oral bacteria to the esophagus and reflux of gastric microbiota. Recognizably, this varies considerably from person to person, even in the apparently healthy population. There have been multiple attempts to classify the healthy esophageal flora into different cluster types. One study evaluated healthy and GERD patients demonstrated two microbiome types, referred to as type I and type II [6]. Type I microbiome associated with healthy subjects, consisted primarily of gram-positive microbes, dominated by those within the Streptococcus genus. Conversely, type II microbiome had a greater presence of gram-negative anaerobes/microaerophiles and primarily correlated with esophagitis (odds ratio, 15.4) and Barrett’s esophagus (odds ratio, 16.5) [6].
Conceivably, the dysbiosis antedates the inflammation. In this hypothesis, there is a notable activation of Toll-like receptors (TLRs) by gram-negative bacterial products compounds produced by gram-negative bacterial, and a subsequent propagation of the inflammatory cascade [7] (Figure 1). Another study further classified esophageal biomes into three community types or clusters: one type dominated by Streptococcus spp., one by Prevotella spp., and another with an intermediate predominance of streptococcus, prevotella, haemophilus and rothia spp [8]. These clusters were associated with a variation in metabolic function. The streptococcus cluster associated with pentose phosphate metabolism, the prevotella cluster was associated with lipopolysaccharide (LPS) production, and, the intermediate cluster was associated with glycolysis and short chain fatty acid (SCFA) production [8]. For all three clusters, progression to reflux-related esophageal disease was associated with increase in relative abundance of gram-negative flora. This supports the TLR/inflammatory cascade hypothesis which is substantiated by the observation that composition of the esophageal microbiome is closely linked to the gastric and oral flora. The prevalent genera including streptococcus, prevotella, haemophilus, fusobacterium, and neisseria, appear similar in both the stomach and oral cavity, suggesting that the oronasal compartment is the primary source of bacteria in the aerodigestive tract [9].
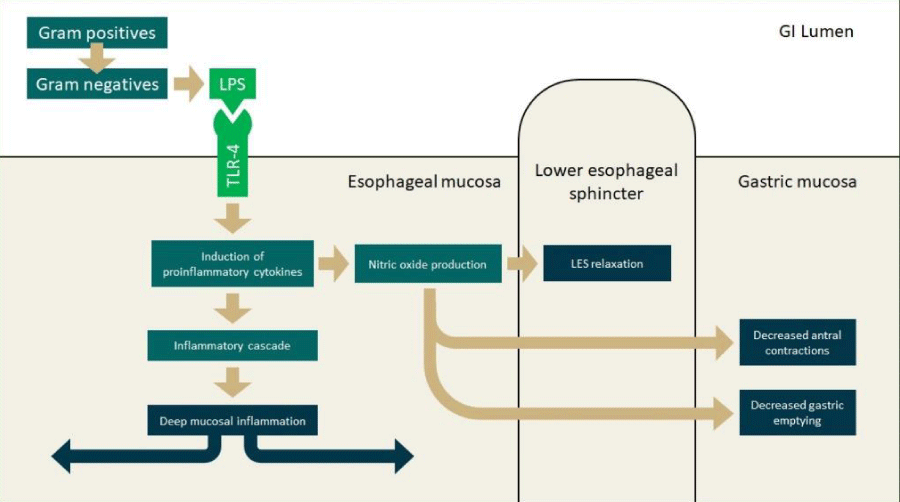
Figure 1: Factors influencing pathogenesis in GERD, Barrett’s esophagus, and esophageal adenocarcinoma. Abbreviations: GERD: Gastroesophageal Reflux Disease; GI: Gastrointestinal; LES: Lower Esophageal Sphincter; LPS: Lipopolysaccharide; TLR: Toll-Like Receptor..
Dysbiosis in disease states
Gastroesophageal reflux disease: It is well recognized that GERD is an inflammatory disease state affecting the lower esophagus related to inappropriate transient relaxation or chronic hypotenisity, of the lower esophageal sphincter. Retrograde reflux of gastric acid with or without bile causes symptoms and inflammatory changes associated with GERD [10]. The most frequent treatment medications are intended to buffer (antacids) or reduce gastric acid secretion are intended using proton-pump inhibitors (PPIs) or histamine-2 receptor antagonists [11]. Untreated GERD may progress and manifest with complications of erosive esophagitis, esophageal stricture, BE, or esophageal adenocarcinoma [11].
Inflammatory pathogenesis of GERD: Histologically, the mucosal inflammation observed in GERD is classically thought to be a consequence of direct chemical injury from gastric acid reflux. The specific factors contributing to epithelial insult were gastric acid and duodenal bile salts. Recent literature, has demonstrated that a concomitant immunogenic pathway exists. It remains unclear however, whether erosive injury or cytokine-mediated inflammation (via response to antigenic bacterial LPS) occurs first. Biopsies of tissue from the distal esophagus in GERD patients frequently show submucosal (cytokine-induced) inflammation, but with intact epithelial cells. These findings are inconsistent with initial surface epithelial chemical injury (erosion). Therefore, a non-erosive mechanism is more likely, and would be the result of immunoreactivity to a foreign biomaterial. In this case, the literature implicates a pathway involving LPS, a well-recognized cell wall constituent of gram-negative bacteria. This LPS is critical to maintain the bacterial cell integrity and viability, but also is a potent inducer of inflammatory responses and can directly modulate the immune system and susceptibility to disease [12]. The interaction of bacterial LPS and TLR-4 directs the innate immune system via NF-κB and cytokine cascade initiating the inflammatory response [12].
There are however, also many protective factors against an inflammatory response, including the stratified squamous epithelial barrier, paracellular adhesion and intracellular buffering. Bypassing or overwhelming these protective mechanisms leads to cellular injury and an inflammatory cascade [10]. In vitro and in-vivo exposure of lower esophageal keratinocytes to acidified-bile salts such as those from duodenogastric reflux promotes local cytokine production and migration of lymphocytic cells, primarily T-cells [13]. Progression of exposure leads to inflammation of the mucosa but preservation of the epithelial cell layer, implying that the main insult to the mucosa is deep rather than superficial. This suggests that the pathogenesis of reflux esophagitis is driven more by the innate immune system than by chemical insult. A study examining biopsies from patients with GERD before treatment with PPIs confirmed histologic damage (proliferation of T-cells, hyperplasia of basal cells, and papillary elongation) without damage to surface epithelial cells [14].
Various inflammatory mediators produced by the mucosa also contribute to lower esophageal sphincter (LES) relaxation. Production of interleukin (IL)-8, among other induced factors (including transient receptor potential channel vanilloid subfamily member-1, substance P, calcitonin gene related peptide, and platelet activating factor) stimulates migration of immune response cells. These factors also induce further IL activation and subsequent NADPH oxidase production of hydrogen peroxide. This peroxide effect on local smooth muscle leads to LES relaxation [15]. It is also hypothesized that nitric oxide, another downstream result of cytokine-induced inflammation, is responsible for the relaxation of the LES and decreases gastric emptying [16]. The production of IL-8 is also inhibited by PPIs through a mechanism involving nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein-1 [17]. This may be a contributing factor to the therapeutic effect of PPIs in GERD.
Role of the microbiome in GERD: Recognizably, LPS is the most abundant and important cell wall constituent of gram-negative bacteria. It is vital for bacterial cell integrity, viability, and defense against environmental stress [18]. The TLR-4 protein site in humans is the best characterized of several sensing receptors which mediate LPS-induced signal transduction. Following disruption of the epithelial barrier, increased LPS-TLR-4 binding activates production of IL-18, which induces a cascading inflammatory response.7 This activation of TLR-4 is pivotal for both infectious and noninfectious (e.g. allergic or autoimmune) related inflammation and is a major mechanism for pathogenesis of inflammatory disease states by gram-negative flora [19]. Further TLR-based signaling promotes transcription of pro-inflammatory chemokines, including IL-1, IL-6, IL-8, and tumor necrosis factor-alpha (TNF-α) and mediators such as nitric oxide synthase [20].
Used as a standard for inflammation in models of septic shock, LPS (aka endotoxin) is an intrinsic cell membrane component in gram-negative bacteria. It is a potent pathogen-associated molecular pattern (PAMP) which alerts the host immune system that something non-self is present [21]. This occurs first by interaction with TLR-4, which recognizes the PAMP, and signals cells to produce a series proinflammatory cytokines, in particular, TNF-α and interferon (IFN)-γ [12]. The former induces nitric oxide synthase [22] and the latter affects the sensitivity to LPS [23]. The subsequent release of NO triggers both vasodilation and potentiates response by activated macrophages [24]. In smaller amounts, LPS triggers a localized inflammatory response. When the concentration of LPS is high, as it would be in septic shock, hypotension, thrombosis, and mortality can occur. Additional mechanisms for the recognition of LPS including TLR-2, TRPV1 and 2, and others are being investigated [25].
Chemokines also lead to production of nitric oxide and cyclooxygenase (COX)-2, which promote lower esophageal sphincter relaxation and delayed gastric emptying, respectively [26]. This is exemplified in literature discussing campylobacter spp., specifically Campylobacter concisus, which is observed frequently in patients with esophageal pathology [27]. C. concisus, a common oral flora, is not often found in the healthy esophagus. However, colonization by the organism was observed at the site of histologic changes due to GERD and BE as well as inflammatory bowel disease [28]. Additionally, there is a strong correlation between C. concisus and an increased level of the proinflammatory cytokine IL-18 [27,29].
The focal effects of bacterial colonization suggest a local bacterial adherence, facillitated by a biofilm formation. Biofilm, develops via a structurally organized community of flora that stimulate local microbial secretion of an environmentally protective coating (frequently e.g. mucopolysaccharide, extracellular polysaccharide ,dextran and adhesion molecules) [30]. Biofilms have been observed in association with GI disease, most notably in oral and colonic pathology [31,32]. Biofilm-associated proliferation may present a framework for understanding esophageal pathology although further research regarding the composition the composition of native flora, and their three-dimensional organization is needed [27]. Bacteria behave quite differently in bulk culture compared to when they are in an established biofilm. Among other things, they are much harder to kill. Individual microbes are also much closer together than typical, which changes the quorum threshold and ultimately their behavior, including upregulation of bacteriocin synthesis [33].
Therapeutic regimens, especially PPIs, have been demonstrated to alter both the gastroesophageal as well as colonic microbiomes. The use of PPIs in GERD patients has been demonstrated to affect diversity of gastric, esophageal, and fecal flora, mainly by allowing more proximal organisms to populate more distal areas [34-36]. It is unclear if this is a protective or injurious effect although this could be a contributing factor for increased infection risk and may play a role in the association with fecal microbiome-related disease such as clostridium difficile infection [37]. Reduced diversity, (e.g. chemotherapy, radiation, antibotics) promotes the proliferation of C. difficile. The spores (typical to all Clostridium spp.) are able to survive when other bacteria (that would have otherwise inhibited C. difficile proliferation) have been eradicated.
It has been theorized that metabolic activity of the colonic biome may further contribute to GERD progression [38]. Colonic breakdown of fermentable oligo-, di-, monosaccharides and polyols (FODMAPs) produces SCFAs, some of which contribute to LES relaxation [39,40]. A common FODMAP, fructooligosaccharides have been demonstrated to increase the frequency of transient lower esophageal sphincter relaxations (TLESRs) and reflux episodes [39]. The mechanism for this may be through stimulation of Peptide YY (PYY) production or GLP-1 production [39,41]. Some SCFAs as well as PYY act to inhibit gastric motility as well as to relax lower esophageal sphincter tone. The result is retention of gastric contents as well as susceptibility to reflux, which has implications for dietary intake and colonic microbiome as well as motility alteration within the scope of GERD [40]. These SCFAs, however, do not all have the same effect (at least on the motility of the guinea pig colon). Butyrate increases frequency of propulsive colonic contraction, whereas propionate and acetate decrease this frequency [42].
Barrett’s esophagus
Barrett’s esophagus is an intestinal metaplasia of the distal esophageal epithelium characterized by transition from normal stratified squamous epithelial composition to columnar mucosa [43,44]. Development is thought to be in response to chronic inflammation of the mucosa secondary to gastroesophageal reflux. The associated risks for esophageal adenocarcinoma (EAC) are well recognized [6,26,45].
Inflammatory pathogenesis of BE: As seen in the pathogenesis of GERD, expression of proinflammatory cytokines such as IL-1B, IL-6, and IL-8 is linked to the transition to metaplasia seen in BE. A mouse model of BE demonstrated that IL-1B and IL-6 are overexpressed at the squamocolumnar junction of the esophagus and promote inflammation of gastric cardiac stem cells [46]. The inflammation provides a further pathway for progression to dysplasia.
Another mouse model of Barrett’s esophagus has revealed that a high fat diet (HFD) is associated with esophageal dysplasia through alteration of the microbiome [47]. The HFD increases production of IL-8/C-X-C Motif Chemokine Receptor 1 chemokines, known to be upregulated in a pro-inflammatory state, and these stimulate the migration of immature granulocytic cells in the esophagus, promoting local inflammatory responses [47].
With the increased abundance of gram-negative species in the distal esophagus, the related increase in LPS may trigger a pro-inflammatory response through an increased release of chemokines/cytokines. The host’s response to the increased level of LPS (intrinsic to gram-negative organisms) results in NF-κB activation of the epithelial cells [48]. The NF-κB molecular pathway serves as an initial response step to noxious stimuli (chemical, bacterial, and viral) and assumes a role of upregulating inflammation, innate immune responses, adaptive responses, apoptosis inhibition, cell proliferation and differentiation. IL-1B and IL-8 are increased as a result of NF-κB activation and the secreted cytokines create a positive feedback loop eliciting a more robust innate immune response in BE [26].
Role of the microbiome in BE: As seen in patients with GERD, patients with BE have a distinct microbiome composition [49]. Type II microbiomes, as described earlier, are associated with progression of GERD to BE [50]. These type II microbiomes demonstrate a decrease in Streptococcus spp. and an increase in gram-negative anaerobes/microaerophiles including Veillonella, Prevotella, Haemophilus, Neisseria, Rothia, Granulicatella, Campylobacter, Porphyromonas, Fusobacterium, and Actinomyces spp [6]. This transition from gram positive to gram negative relative abundance is thought to be associated with the pathogenesis of esophagitis as well as the metaplastic progression to BE [50,51]. Expression of LPS from gram-negative bacteria, and subsequent activation of the TLR-4-NF-κB pathway is associated with expression of IL-8 and COX-2. The levels of both are directly correlated with transition from metaplasia to dysplasia [52]. It is also possible that gastric acid could contribute to conversion from type I to type II microbiome by killing acid-sensitive bacteria in the esophagus [53]. Gram-negative organisms are more acid-resistant, and in many cases highly tolerant of bile, which is toxic for gram-positive organisms [54,55].
Molecular products secreted by these flora or components of the bacterial wall such as LPS interact with TLRs and continue the inflammatory cascade seen in reflux esophagitis, preventing resolution of mucosal changes. During the ongoing inflammatory process, changes in the local flora predispose the local squamous epithelial tissue towards metaplasia to columnar epithelium. Wild-type murine models given a HFD demonstrated increase in goblet cell prevalence and relative neutrophil presence compared to germ-free models [47]. This suggests that local microbe-epithelium interactions, presumed to be through microbe product-TLR binding, is a possible mechanism for the metaplastic process. Furthermore, there may be a role of the colonic microbiome in this process as an increase of the Firmicutes:Bacteroidetes ratio within the colon is seen in BE [47]. Many firmicutes metabolize FODMAPs and dietary fiber into SCFAs. As previously described, SCFA production is associated with decreased LES tone as well as decreased gastric motility through stimulation of PYY mediated smooth muscle effects [40].
There is considerable overlap between periodontal and esophageal flora, with a similar firmicutes/bacteroidetes ratio [56]. It is presumed that this similarity in composition is mainly from movement of microbes in a distal direction. A recent study found a distinct oral microbiome that is associated with presence of BE [49]. This case-control study analyzed the oral microbiome using a three-taxon model, (lautropia, streptococcus, and unspecified genus in the order of bacteroidales) which distinguished the microflora of patients with BE from healthy individuals. Albeit a small number of patients, this model of clustering predicted patients with BE with a sensitivity of 97% and specificity of 88% [49]. The model suggests decreased abundance of Lautropia and increased abundance of streptococcus and enterobacteriaceae in BE patients. The increased presence of Lautropia and Enterobacteriaceae, both gram negative, correlates with the LPS/TLR-4 hypothesis, but increased abundance of Streptococcus spp. suggests that pure gram-positive/negative ratio may provide an incomplete explanation. More broadly, increased firmicutes suggests that SCFA production may also play a role in BE pathogenesis. Larger investigations are needed to further evaluate the role of oral dysbiosis and the diagnostic implications for BE because environmental factors can influence the oral microbiome [57].
Esophageal cancer
Esophageal cancer is growing in incidence in many areas of the world [58,59]. Squamous cell carcinoma and adenocarcinoma comprise the two major histological subtypes [60]. Globally, esophageal squamous cell carcinoma (ESCC) is the more common of the two, making up approximately 88% of esophageal cancers [60,61]. Risk factors for both malignancies overlap, although etiologies and incidences vary [62].
Inflammatory pathogenesis
The etiology of EAC is associated with long standing inflammation or mucosal injury, such as that seen in reflux esophagitis. The pathophysiology contributing to this disease state is complex involving an interplay between environmental factors, genetic susceptibilities, and host dynamics. Approximately 80% of cases can be attributed to GERD, cigarette smoking, obesity, and low fruit and vegetable consumption [63]. GERD is one of the strongest risk factors for EAC, correlating strongly with duration and frequency of symptoms. Other risk factors include BE, motor disorders of the esophagus, other malignancies, medications, and environmental exposures [64]. BE is the risk factor that has the strongest association with EAC with estimates that BE progresses to high-grade dysplasia at a rate of 0.5% to 0.9% per year [65]. However, EAC, like the complications of GERD discussed above, can arise without preceding symptoms. As discussed previously, the pathogenesis of GERD follows an inflammatory-mediated cascade rather than through direct surface mucosal chemical injury. Conversely, GERD is not a risk factor for ESCC.
Recent epidemiological studies have observed that the incidence of EAC to be on the rise in the Western world, with a 6-fold increase in the United States alone [66].The recent rise in incidence in the Western world suggests an environmental etiology at play. Studies have explored factors such as diet, smoking, obesity, H. pylori infection, and antibiotics [53].
Squamous cell carcinoma: Esophageal squamous cell cancer is a complex disease, with many predisposing factors, involving both genetic as well as environmental components. Incidence of the disease is influenced by environmental exposure, but there is regional variation to the nature of exposure. The highest areas of incidence include East Asia, Southeastern Africa, and Southeastern South America [61]. Smoking and tobacco use are the most common risk factors in wealthier populations, while hot-liquid consumption, dietary carcinogens, and poor dentition contribute enough to incidence that poverty itself is a risk factor for pathogenesis [61,67,68]. There is a growing body of literature evaluating genetic susceptibility that may increase carcinogenesis following an environmental trigger [61].
Heavy alcohol consumption is a strong risk factor for ESCC. Acetaldehyde, a direct metabolite of ethanol oxidation, inhibits DNA repair through a variety of mechanisms. Ethanol may also directly induce production of reactive oxidation species and promotes aberrant epigenetic modification, in particular DNA methylation [69]. Abnormal methylation of genes associated with carcinogenesis inhibits expression of tumor-suppressor genes, promotes oncogene transcription, and is a the major proposed mechanism for the direct carcinogenic effect of ethanol [70].
Pathogenesis of ESCC is associated with overexpression of inflammatory mediators. Persistent production of NF-κB and activation of TLR-4 have both been demonstrated to be present in early stage ESCC whereas production/activation decreases with progression to advanced stages [71]. TLR-4, in particular, activates an innate inflammatory response with subsequent activation of an acute to chronic inflammatory cascade [72]. This suggests that the presence of external factors affect the local mucosa-microbe interaction. This leads to a localized inflammatory reaction. The persistence of this inflammation, coupled with a genetic predisposition, triggers hyperproliferation of squamous tissue and progression to carcinoma.
Role of the microbiome in esophageal cancer
There is increasing literature evaluating the relationship between the microbiome and esophageal cancer [73]. The microbiome is altered in precursors to esophageal carcinoma, such as the abnormal type II microbiomes enriched with gram-negative bacteria that are mainly associated with GERD and BE (Table 1). This alteration of the microbiome is potentially involved in carcinogenesis. The microbiota of cancerous esophageal tissue has been characterized to be profoundly affected by the oral microbiome, and periodontopathic species derived from the oral cavity. Oral microbial composition has been associated with the risk of EAC and ESCC [73]. In fact, many of the studies implicate oral bacteria in the etiology of oral, esophageal, gastric and other GI cancers [74]. Due to bacteria migration, the oral and gastric microbiota shape the esophageal microbiome and therefore may contribute to esophageal carcinogenesis [73]. However, there are variations in microbiota even between ESCC and EAC.
| Table 1: Comparison of parameters of inflammatory pathogenesis and microbiome changes seen in GERD, Barrett’s esophagus, and esophageal cancer. | ||
| Inflammatory Pathogenesis | Role of the Microbiome | |
| GERD | Initally,esophageal bacterial dysbiosis activates a localized submucosal cytokine inflammatory response leading to subsequent direct chemical damage by acid/bile. | Associated with transition from to Type I to Type II microbiome (more gram-negative anaerobes/microaerophiles). Interaction of LPS with TLR-4 activates NF-кB and subsequent cytokine cascade. |
| Barrett’s Esophagus | Following presence of GERD and continued local expression of interleukins such as IL-1B and IL-6. | LPS from gram-negative bacteria activates the TLR-4-NF-кB pathway and increases IL-8 and COX-2 expression. Both IL-8 and COX-2 are associated with progression of metaplasia to dysplasia. |
| Esophageal Cancer | EAC: GERD and Barrett’s esophagus increase risk due to ongoing mucosal inflammation. ESCC: Environmental factors such as tobacco and alcohol use increase local inflammation and risk of malignant transformation. |
EAC: Associated with specific bacteria, carcinogenic role of cytokines via TLR pathways, COX-2 isoform increase. ESCC: Decreased microbial diversity, carcinogenic role of cytokines, environmental factor alterations (e.g. alcohol, tobacco) alter biome and microbe-immune crosstalk. |
| Abbreviations: COX: Cyclooxygenase; EAC: Esophageal Adenocarcinoma; ESCC: Esophageal Squamous Cell Carcinoma; GERD: Gastroesophageal Reflux Disease; IL: Interleukin; LPS: Lipopolysaccharide; NF-Кb: Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells; TLR: Toll-Like Receptor | ||
Adenocarcinoma: The microbiome of EAC has been characterized as similar to the type II microbiome, but individual periodontopathic species derived from the oral microbiome are found to be associated with disease: treponema denticola, streptococcus mitis, and streptococcus anginosus [75]. The latter two are gram positive and this suggests a different pathway involving migration or SCFA metabolism, as described in the BE pathogenesis rather than the previously discussed LPS/dysbiosis pathway. This suggests that periodontitis and inadequate oral hygiene may be associated with increased esophageal cancer risk [76]. In particular, fragments of s. anginosus have been isolated in head and neck carcinomas as well as in early dysplastic changes of esophageal and gastric cancer [77]. This implies that s. anginosus is associated with numerous malignancies of the upper digestive tract. The exact mechanism underlying this process has not been delineated. However, induction of inflammatory cytokines by infection with s. mitis and s. anginosus has been demonstrated [75]. Other periodontal species have been associated with esophageal cancer [73].For instance, it has been found that the periodontal pathogens tannerella forsythia, veillonella, selenomonas, and treponema spp. are associated with higher risk of EAC [78,79].
Decreased streptococcus prevalence is associated with a lower risk of EAC implying that specific flora are inversely related to malignant transformation [73]. Further evidence of this inverse correlation is provided by lower EAC risk is associated with H.pylori infection [73]. In one study, the microbiome in both normal subjects and EAC was characterized to be more alike than to BE comparisons, with an increased relative abundance of bifidobacteria, bacteroides, fusobacteria, veillonella, staphylococcus and lactobacilli and decreased relative abundance of campylobacter when compared with BE samples [27]. Other protective factors such as bacterial biosynthesis of carotenoids by oral neisseria spp. were also associated with protection against EAC [73]. This suggests that commensal colonization by specific flora may be protective from the metaplastic process by inhibiting proliferation of pro-inflammatory flora as well as through synthesis of vitamins or perhaps by eliciting a generally protective immune response.
Increased EAC risk has also been associated with depletion of certain bacteria. For instance, depletion of the commensal genus neisseria and the species streptococcus pneumoniae are associated with higher EAC risk [73]. This is corroborated by other studies wherein microbial diversity has also been shown to be decreased [79,80]. It has been postulated that once carcinogenesis has begun, streptococci leave the local environment to invade surrounding tissue [81]. Conceivably, etiology from GERD to BE to EAC is a progression of microbiotas. Each stage opens the door to a new microbiota behavior, for example, migration into tissue and cells. Thus, a “snapshot” assessment and ccomparison at any one time, may not be all encompassing and truly reflective of the entire progressive changes.
Toll-like receptors are a class of proteins that play a key regulatory role within the innate immune system. A potential mechanism by which the microbiome participates in carcinogenesis, is via TLRs [79]. TLRs 1-3, 6, 7, and 9 are significantly upregulated in EAC [81]. Both TLR-4 and TLR-5 have also been suggested as potential mediators of the progression from reflux disorders to EAC. This suggests an association between the TLR signaling pathway and the altered microbiome. In tissue biopsies from the esophagus, TLR-4 (whose natural ligand is LPS) expression is significantly increased in EAC and BE when compared to normal esophagus [53]. Further, activation of the TLR-4-NF-κB pathway is evident in reflux disorders and may contribute to malignant transformation [26]. Therefore, with abnormal type II microbiomes, there is an increased relative abundance of gram-negative bacteria, over resultant stimulation of TLR-4. This may iniate a larger may trigger a larger and more carcinogenic inflammatory cascade. Further, expression of the COX-2 isoform, an LPS-TLR-4-NF-κB pathway downstream gene, is elevated in esophageal carcinomas. It has been found that there is an increase of COX-2 that occurs along the progression from low-grade dysplasia to high-grade dysplasia in the EAC pathway [82]. This implies that the activation of the LPS-TLR-4-NF-κB pathway may contribute to malignant transformation. The theory has been experimentally tested in a murine model wherein the presence of E. coli induced activation of TLRs implicated in EAC [81].
Squamous cell carcinoma: As with esophageal adenocarcinoma, ESCC has been shown to be associated with periodontal pathogens and poor oral hygiene [61,72]. Specifically, the abundance of the periodontal pathogen Porphyromonas gingivalis trended with higher risk of ESCC [83]. In one study, poor oral health was reported as a risk factor for esophageal squamous dysplasia [84]. To this point oral SCC, also associated with poor oral hygiene, has been linked to changes in the oral microbiome (firmicutes, streptococcus, actinobacteria, and rothia), which were substantially decreased in relation to normal tissue [85]. Oral SCC has been shown to be accompanied with other squamous cell carcinomas of the digestive tract [86]. It has been suggested that a region of epithelial cells can be affected by carcinogenic alterations [87].
Subjects with ESCC have also been shown to exhibit decreased microbial diversity [72]. Interestingly, this decrease in microbial diversity has been replicated in other anatomical sites of the GI system such as the stomach with gastritis [74]. and the colon with colorectal cancer [88]. Gastric microbiota changes have also been associated with ESCC, and Clostridiales and Erysipelotrichales orders have been particularly implicated [89].
In addition to direct carcinogenic effects, environmental factors such as alcohol and tobacco may alter the local microbiome and contribute to carcinogenesis. Consumption of alcohol in patients with oral microflora abundant with oxidizing flora leads to production of acetaldehyde, and subsequent DNA repair inhibition, leading to increased susceptibility to oral squamous cell carcinoma [90,91]. Tobacco use is associated with increased abundance of Streptococcus spp. and yeast capable of metabolizing alcohol to acetaldehyde, as well as inhibiting acetaldehyde breakdown, suggesting that persistence of salivary aldehyde can contribute to esophageal carcinogenesis [92]. Alcohol use is associated with an increased firmicutes: bacteroidetes ratio in murine fecal samples [93]. This increased ratio alters local nutrient metabolism and increases serum LPS levels, and may suggest further means of potentiating pathogenesis.
Alcohol may also influence both local and systemic response to microbe-immune crosstalk. Locally, ethanol inhibits epithelial cell expression of tight junction-associated proteins, zonula occludens-1 and claudin-1, increasing barrier permeability and susceptibility to the LPS-mediated inflammatory response [94]. Systemically, both heavy acute and chronic ethanol consumption may also decrease clearance of LPS from the bloodstream, potentiating a systemic pro-inflammatory effect [95].
Esophageal dysmotility
Inflammatory pathogenesis: Mucosal inflammation leading to altered neuromotor function and GI dysmotility has been described in esophagitis and ulcerative colitis [96]. In patients with GERD, an increase in cytokines and chemokines such as IL-1B, IL-6, IL-8, IL-10, interferon-γ, monocyte chemoattractant protein-1, and Regulated-upon-Activation, Normal-T-Cell-Expressed-and-Presumably-Secreted (RANTES) are observed [97]. IL-6, a cytokine released as a result of mucosal damage, affects the circular smooth muscle cells in the lower third of the esophagus and ultimately disturbs muscle contractility. Although the exact mechanism of cytokine effects on afferent neural pathways are unknown, it is proposed that increased cytokine production occurs as a consequent response to gastric refluxate exposure or the dysbiosis/LPS evident with GERD. These cytokines, including IL-6, are able to alter normal esophageal contractility [98].
Role of the microbiome in esophageal dysmotility: While there are no studies directly evaluating the role of the local microbiome in the pathogenesis of esophageal motility disorders, there are some investigations that have characterized the microbiome in achalasia, Chagas disease and connective tissue connective tissue diseases such as systemic sclerosis [99]. Additionally, a large volume of literature characterizes the effects of local flora on smooth muscle elsewhere along the GI tract, particularly the colon.
Although Chagas disease is a well-recognized consequence of a tropical parasitic infection caused by Trypanosoma cruzi, samples of flora grown from patients with chagas related megaesophagus has demonstrated a predominance of nitrite- and nitrate-reducing bacteria [100]. Further investigation of patients with achalasia and megaesophagus demonstrate an overgrowth of streptococcus spp., many of which may act as nitrite-/nitrate-fermenters [101]. As previously described, type I esophageal microbiomes are typically seen as ‘normal’ flora and consist primarily of Streptococcus spp. It is possible that the decrease in interaction between esophageal and gastric flora in various megaesophagus states leads to overgrowth of these type I flora, although this has not clearly been characterized. Furthermore, increase in nitrite-/nitrate- fermentation, coupled with increased esophageal retention of food products may contribute to development of squamous cell carcinoma and may explain the increased risk that is seen in these patients [64, 100].
As described previously in relation to GERD and BE, an increase in the fecal firmicutes/bacteroidetes ratio corresponds with an increase in firmicutes that produce SCFAs. These SCFAs and downstream PYY decrease the contraction of gastric smooth muscle [40]. This SCFA-mediated effect following changes in the colonic firmicutes:bacteroidetes ratio has also been demonstrated in cases of decreased colonic motility leading to constipation [102]. While their effect in the terminal ileum appears to be mainly stimulation of peristalsis following ileocolic reflux, effects elsewhere in the GI tract manifest as a decrease in motor tone [103]. More investigation is needed to isolate the effect of SCFA production on esophageal peristalsis.
Connective tissue diseases are associated with gut dysbiosis [104].Most notably, systemic sclerosis, which can lead to fibrosis of the muscular layers of the GI tract and subsequent dysmotility, most frequently manifests in the esophagus [99]. Multiple studies investigating compositional changes in colonic flora have demonstrated decrease in commensal flora such as bacteroides, clostridium, and faecalibacterium, and an increase in potentially invasive flora, such as fusobacterium and desulfovibrio [105-108]. While the mechanism for dysbiotic contribution to fibrosis still requires more investigation, it is believed that epithelial barrier dysfunction leading to microbial inflammatory cascade activation may play a role [108].
Eosinophilic esophagitis
Eosinophilic esophagitis (EoE) is a chronic immune mediated disorder that is characterized endoscopically by fixed esophageal rings, esophageal narrowing, and mucosal friability and diagnosed by biopsy findings of eosinophilic infiltration of the esophageal mucosa [109]. This is frequently associated with atopic disease, most frequently in male patients. Inital treatment once, confirmed not PPI responsive, is with topical steroids and dietary modification to remove potential allergens [109].
Inflammatory pathogenesis: EoE is a multifactorial disease, with several genetic components, frequently atopic condition associations, as well as environmental factors that are presumed to affect pathogenesis. The role of these various factors in development of EoE is still being characterized, and there are several explanations for its inflammatory origins. One theory is that repeat allergen exposure in susceptible individuals may contribute to eosinophil-driven inflammation [110]. This is highlighted in some data that demonstrates childhood PPI use with EoE as theoretically decreasing allergen digestion and prolonging exposure [111].
The progression of EoE is believed to follow genetic or environmental disruption of the epithelial barrier exposing the underlying mucosal tissue to local allergens and bacterial products. These products stimulate secretion of IL-1, IL-8, and migration of T-helper 2 (Th2) cells, which produce IL-5 and stimulate recruitment and activation of eosinophils, as well as IL-13, which stimulates downregulation of desmoglein-1, a cell adhesion molecule integral to the maintenance of the epithelial barrier [112,113]. Local activation of eosinophils leads to toxic degranulation that further stimulates local inflammation and transforming-growth factor (TGF) expression, which mediates structural protein deposition and leads to the characteristic endoscopic and histologic fibrotic findings [112,114].
Role of the microbiome in EoE: Esophageal mucosal eosinophilia and its associated diseases are characterized by a change in the local microbiome [115]. It has been reported that compared to healthy controls without EoE, the esophageal microbiome of patients with active EoE contained a significantly greater abundance of Haemophilus spp [115]. Further, this change was reversed by following standard treatment, in-essence, restoring the bacterial composition to one more typical of GERD and healthy subjects. Additionally, the bacterial load, but not the diversity, was increased in subjects with EOE and GERD relative to healthy controls [115]. However in another study, patients with active EoE had a significantly greater abundance of Neisseria and Corynebacterium spp. when compared with controls (restored EoE patients, now apparently healthy) [116], haemophilus and neisseria are both genera within the proteobacteria phylum and are associated with activation of the inflammatory cascade [117].
Composition of more proximal flora may also contribute to pathogenesis of EoE. A study of the salivary microbiome in pediatric patients demonstrated significant differences in bacterial composition in EoE patients compared to non-EoE controls [118]. Specifically, there was an association with haemophilus spp. with active disease, similar to the changes seen in esophageal samples. This suggests a close interaction between the flora of both locations, and that characterization of salivary dysbiosis may be a surrogate marker for EoE disease activity [115,118].
Role of bacteriocins
Bacteriocins are small peptide molecules that are expressed by bacteria in response to stress. Their primary role is presumed to be inhibition of competitive local flora as well as newly introduced foreign competitors for the same niche. This may explain why most studies of probiotic consumption observe transient effects and lack of long-term colonization. They have been traditionally used with the food industry internationally for preservation [119]. They may also play a larger role in regulation of the microbiota within the human GI tract. These bacterial products are ribosome derived peptides typically classified by the nature of bactericidal activity (target and potency), amino acid sequence and peptide structure (including post-translational modifications), bacteriocin operon, molecular weight, and charge, and are separated into four classes [120-122].
Bacteriocins are potent antibacterial agents against very specific strains of bacteria. Exceptions do occur, however, and broad-spectrum antibacterial peptides are known, and use is made of them as preservatives for food (e.g. nisins and pediocins). Several bacteriocins have been noted to be exquisitely toxic to human pathogens, both gram positive, (e.g. MRSA) and gram negative, (e.g. listeria monocytogenes) [123,124]. These inhibitory effects tend to add resilience to the established composition of the local flora.
Another potential role of bacteriocins is in their antineoplastic effect. Research studying the effect of nisin Z on head, neck, and squamous cell carcinoma (HNSCC) has found that bacteriocins may trigger apoptosis in vitro, and have been demonstrated to reduce the size of tumor xenografts [125,126]. Studies have also observed that such bacteriocins appeared benign to normal healthy cells at the concentrations that were effective against the targeted cancer cells. However, greater study of the cytotoxic effects of bacteriocins is needed. Their role in inhibition of carcinogenesis more generally is also still developing [119].
Implications for therapy
Prebiotics: There is increasing evidence that dietary intake has a profound effect on microbiome balance and activity. This effect plays a key role in reducing dysbiosis related induction of inflammatory signaling cascades [127-129]. As the pathogenic role of esophageal dysbiosis microbiota becomes more clear, targeted therapeutic intervention using specific prebiotics appears promising- specifically when focused on enhancing the gram-positive/gram-negative ratio. It may be possible, by selectively promoting the activity of certain organisms that produce metabolites, proteins or peptides, that a local microbiome may be curated to a more favorable state. Isomaltooligosaccharides have been demonstrated to increase the number of gram-positive flora, especially bifidobacterium and lactobacillus spp. Prebiotics, including Maltosyl-isomaltooligosaccharide (MIMO), are aimed at improving gram-positive/gram-negative ratio in the esophagus. This type of intervention has demonstrated some potential in reducing or eliminating GERD symptoms in case series [130].
Probiotics: The use of probiotics to modify the gut microbiome has been studied in a variety of GI disease states, and there are several investigations into usage to decrease GERD symptoms. Probiotic formulations that include strains mainly within the genera lactobacillus and bifidobacterium have been studied and have demonstrated reduction in symptoms when used as monotherapy [131-134]. However, many studies are limited by quality due to limitations in experimental design such as producing an adequate placebo for control [135]. A randomized-controlled study utilizing bacillus subtilis and enterococcus faecium with PPI demonstrated decrease in diarrhea symptoms and small intestinal bacterial overgrowth but did not reduce GERD symptoms or healing rate compared to PPI alone [136]. Additionally, recent evidence demonstrated that although orally administered probiotics can remain viable, there may be a marked resistance by the host to mucosal colonization [137].
Antibiotics: Antibiotics are also a possible therapeutic option for modifying the microbiome, given their efficacy in the treatment of GI infectious diseases. While routine use of antibiotics for the treatment of esophageal disease has not been investigated, it has been used with success elsewhere in the GI tract for dysbiosis related disease states. In patients with small intestinal bacterial overgrowth (SIBO), a condition characterized by proliferation of commensal flora within the small bowel, antibiotics with poor oral bioavailability such as rifaximin have been used with efficacy, though data on the use of systemic antibiotics is limited [138]. Given the implications of emerging antibiotic resistant pathogens and risk for c. difficile colitis, it seems unlikely that this approach will be viable for esophageal diseases [139,140].
Bacteriocins: Another possible avenue for intervention involves application of a bacteriocins, whether directly or indirectly, via selectively providing prebiotics to specific organisms. As previously mentioned, bacteriocins have a potential use through two mechanisms: (1) direct antibiotic effect as well as (2) cytotoxic effect towards neoplastic cells.
While this is an active area of research, and inhibitory isolates that target methicillin-resistant staphylococcus aureus [123] and vancomycin- resistent enterococcus [141]. have been isolated, more research is needed regarding products that target specific organisms as well as subsequent literature to evaluate for efficacy and safety [141].
Bacteria have been found within tumors exhibiting cooperative activity with cancer cells—they can metabolically deactivate chemotherapeutics (gemcitabine) and induce chemotherapeutic resistance in pancreatic cancer [142]. A direct therapeutic bacteriocin that could disable these organisms with minimal cytotoxicity to healthy cells could have great potential as adjuvant therapy.
Bacteriocins have two potential weaknesses as potential therapeutic agents:
1. Cytotoxic effects on non-target tissues.
2. Targeted pathogenic species to become bacteriocin resistant.
Fortunately, one promising solution involves improvements in targeted drug delivery via encapsulation or attachment of bacteriocins to macromolecule-based, metal, or polymer-based nanoparticles [143]. This has the added potential for modulating or increasing intended effect through attachment of adjuvants to the same nanodelivery particles.
Dysbiosis of the local flora within the esophagus is associated with progression to various gastroesophageal disease states including GERD, BE, EAC, ESCC, esophageal dysmotility and EoE. Dysbiosis may be caused by multiple factors including diet, lifestyle, antibiotic use, and/or food-borne pathogens and remains to be further studied. At present, prebiotics and perhaps direct bacteriocin therapies, have the most promising new potential for treatment of common diseases of the esophagus.
- Peery AF, Crockett SD, Murphy CC, Lund JL, Dellon ES, et al. Burden and Cost of Gastrointestinal, Liver, and Pancreatic Diseases in the United States: Update 2018. Gastroenterology. 2019;156: 254-272.e11. PubMed: https://pubmed.ncbi.nlm.nih.gov/30315778/
- Almario CV, Ballal ML, Chey WD, Nordstrom C, Khanna D, et al. Burden of Gastrointestinal Symptoms in the United States: Results of a Nationally Representative Survey of Over 71,000 Americans. Am J Gastroenterol. 2018;113: 1701-1710. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6453579/
- Hayeck TJ, Kong CY, Spechler SJ, Gazelle GS, Hur C. The prevalence of Barrett’s esophagus in the US: estimates from a simulation model confirmed by SEER data. Dis esophagus Off J Int Soc Dis Esophagus. 2010;23: 451-457. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2896446/
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020; 70: 7-30. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31912902
- Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Heal Dis. 2015; 26. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25651997
- Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and Intestinal Metaplasia of the Distal Esophagus Are Associated With Alterations in the Microbiome. Gastroenterology. 2009;137: 588-597. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2963147/
- Ajayi TA, Cantrell S, Spann A, Garman KS. Barrett’s esophagus and esophageal cancer: Links to microbes and the microbiome. PLoS Pathog. 2018;14: e1007384. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30571768
- Deshpande NP, Riordan SM, Castaño-Rodríguez N, Wilkins MR, Kaakoush NO. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome. 2018;6. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30558669
- Bassis CM, Erb-Downward JR, Dickson RP, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. MBio. 2015; 6.
- Orlando RC. The integrity of the esophageal mucosa. Balance between offensive and defensive mechanisms. Best Pract Res Clin Gastroenterol. 2010 ;24: 873-882. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21126700
- Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol. 2013;108: 308-328. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23419381
- Mazgaeen L, Gurung P. Recent Advances in Lipopolysaccharide Recognition Systems. Int J Mol Sci. 2020; 21: 379. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31936182
- Souza RF, Huo X, Mittal V, Schuler CM, Carmack SW, et al. Gastroesophageal Reflux Might Cause Esophagitis Through a Cytokine-Mediated Mechanism Rather Than Caustic Acid Injury. Gastroenterology. 2009;137: 1776-1784. PubMed: https://pubmed.ncbi.nlm.nih.gov/19660463/
- Dunbar KB, Agoston AT, Odze RD, et al. Association of acute gastroesophageal reflux disease with esophageal histologic changes. JAMA - J Am Med Assoc. 2016; 315: 2104-2112. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27187303
- Harnett KM, Rieder F, Behar J, Biancani P. Viewpoints on Acid-Induced Inflammatory Mediators in Esophageal Mucosa. J Neurogastroenterol Motil. 2010; 16: 374-388. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21103419
- Nikaki K, Sawada A, Ustaoglu A, Sifrim D. Neuronal Control of Esophageal Peristalsis and Its Role in Esophageal Disease. Curr Gastroenterol Rep. 2019; 21: 1-9. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31760496
- Huo X, Zhang X, Yu C, et al. In oesophageal squamous cells exposed to acidic bile salt medium, omeprazole inhibits IL-8 expression through effects on nuclear factor-κB and activator protein-1. Gut. 2014;63: 1042-1052. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3959621/
- Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996; 60: 8-26. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8699127
- Liaunardy-Jopeace A, Gay NJ. Molecular and cellular regulation of Toll-like receptor-4 activity induced by lipopolysaccharide ligands. Front Immunol. 2014; 5: 473. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25339952
- Kany S, Vollrath JT, Relja B. Cytokines in inflammatory disease. Int J Mol Sci. 2019; 20: 6008. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6929211/
- Lewis AJ, Seymour CW, Rosengart MR. Current Murine Models of Sepsis. Surg Infect (Larchmt). 2016; 17: 385-393. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4960474/
- Fonseca SG, Romão PRT, Figueiredo F, Morais RH, Lima HC, et al. TNF-α mediates the induction of nitric oxide synthase in macrophages but not in neutrophils in experimental cutaneous leishmaniasis. Eur J Immunol. 2003; 33: 2297-2306. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12884305
- Salim T, Sershen CL, May EE. Investigating the role of TNF-α and IFN-γ activation on the dynamics of iNOS gene expression in lps stimulated macrophages. PLoS One. 2016; 1: e0153289. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4898755/
- Sharma JN, Al-Omran A, Parvathy SS. Role of nitric oxide in inflammatory diseases. Inflammopharmacology. 2007;15: 252-259. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18236016
- Cauwels A, Bultinck J, De Zwaef R, Vandendriessche B, Magez S, et al. Nitric oxide production by endotoxin preparations in TLR4-deficient mice. Nitric Oxide - Biol Chem. 2014; 36: 36-43. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24269486
- Yang L, Francois F, Pei Z. Molecular pathways: Pathogenesis and clinical implications of microbiome alteration in esophagitis and barrett esophagus. Clin Cancer Res. 2012;18: 2138-2144. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22344232
- Blackett KL, Siddhi SS, Cleary S, et al. Oesophageal bacterial biofilm changes in gastro-oesophageal reflux disease, Barrett’s and oesophageal carcinoma: Association or causality? Aliment Pharmacol Ther. 2013; 37: 1084-1092. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23600758
- Liu F, Ma R, Wang Y, Zhang L. The clinical importance of campylobacter concisus and other human hosted Campylobacter species. Front Cell Infect Microbiol. 2018; 8: 243. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30087857
- Corning B, Copland AP, Frye JW. The Esophageal Microbiome in Health and Disease. Curr Gastroenterol Rep. 2018; 20: 39. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30069679
- Nadell CD, Ricaurte D, Yan J, Drescher K, Bassler BL. Flow environment and matrix structure interact to determine spatial competition in Pseudomonas aeruginosa biofilms. Elife. 2017; 6: e21855. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28084994
- Marsh PD. Dental plaque as a biofilm and a microbial community - implications for health and disease. BMC Oral Health. 2006; 6 (Suppl 1): S14. PubMed: https://bmcoralhealth.biomedcentral.com/articles/10.1186/1472-6831-6-S1-S14
- Dejea CM, Fathi P, Craig JM, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. 2018; 359: 592-597. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29420293
- Lebeaux D, Ghigo JM, Beloin C. Biofilm-Related Infections: Bridging the Gap between Clinical Management and Fundamental Aspects of Recalcitrance toward Antibiotics. Microbiol Mol Biol Rev. 2014; 78: 510-543. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25184564
- Imhann F, Bonder MJ, Vila AV, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016 ;65: 740-748. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26657899
- Jackson MA, Goodrich JK, Maxan ME, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2016; 65: 749-756. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26719299
- Macke L, Schulz C, Koletzko L, Malfertheiner P. Systematic review: the effects of proton pump inhibitors on the microbiome of the digestive tract—evidence from next-generation sequencing studies. Aliment Pharmacol Ther. 2020;51: 505-526. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31990420
- Janarthanan S, Ditah I, Adler DG, Ehrinpreis MN. Clostridium difficile-associated diarrhea and proton pump inhibitor therapy: A meta-analysis. Am J Gastroenterol. 2012; 107: 1001-1010. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22710578
- Uno Y. GERD by colonic fermentation. Neurogastroenterol Motil. 2020; 32.
- Piche T, Des Varannes SB, Sacher-Huvelin S, Holst JJ, Cuber JC, et al. Colonic fermentation influences lower esophageal sphincter function in gastroesophageal reflux disease. Gastroenterology. 2003; 124: 894-902. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12671885
- Ropert A, Cherbut C, Roze C, et al. Colonic fermentation and proximal gastric tone in humans. Gastroenterology. 1996; 111: 289-296. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8690193
- Larraufie P, Martin-Gallausiaux C, Lapaque N, et al. SCFAs strongly stimulate PYY production in human enteroendocrine cells. Sci Rep. 2018; 8.
- Hurst NR, Kendig DM, Murthy KS, Grider JR. The short chain fatty acids, butyrate and propionate, have differential effects on the motility of the guinea pig colon. 2014; 26: 1586-1596. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25223619
- Shaheen NJ, Falk GW, Iyer PG, Gerson LB. ACG Clinical Guideline: Diagnosis and Management of Barrett’s Esophagus. Am J Gastroenterol. 2016;111: 30-50. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27356842
- Naini BV, Souza RF, Odze RD. Barrett’s esophagus: A comprehensive and contemporary review for pathologists. Am J Surg Pathol. 2016;40: e45-e66. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4833583/
- Pei ZH, Yang L, Peek RM, Levine SM, Pride DT, et al. Bacterial biota in reflux esophagitis and Barrett’s esophagus. World J Gastroenterol. 2005; 11: 7277-7283. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3025696/
- Quante M, Bhagat G, Abrams JA, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of barrett-like metaplasia. Cancer Cell. 2012; 21: 36-51. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22264787
- Münch NS, Fang HY, Ingermann J, Maurer hc, Anand a, et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology. 2019; 157: 492-506.e2. PubMed: https://pubmed.ncbi.nlm.nih.gov/30998992/
- Pasparakis M. Role of NF-κB in epithelial biology. Immunol Rev. 2012;2 46: 346-358. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22435565
- Snider EJ, Compres G, Freedberg DE, et al. Barrett’s esophagus is associated with a distinct oral microbiome. Clin Transl Gastroenterol. 2018; 9. 156. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29491399
- Yang L, Chaudhary N, Baghdadi J, Pei Z. Microbiome in reflux disorders and esophageal adenocarcinoma. Cancer J (United States). 2014; 20: 207-210. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24855009
- Liu N, Ando T, Ishiguro K, et al. Characterization of bacterial biota in the distal esophagus of Japanese patients with reflux esophagitis and Barrett’s esophagus. BMC Infect Dis. 2013;13: 130. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23496929
- Konturek PC, Nikiforuk A, Kania J, Raithel M, Hahn EG, et al. Activation of NFkappaB represents the central event in the neoplastic progression associated with Barrett’s esophagus: a possible link to the inflammation and overexpression of COX-2, PPARgamma and growth factors. Dig Dis Sci. 2004; 49: 1075-1083. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15387324
- Neto AG, Whitaker A, Pei Z. Microbiome and potential targets for chemoprevention of esophageal adenocarcinoma. Semin Oncol. 2016; 43: 86-96. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26970127
- Nojoumi SA, Smith DG, Rowbury RJ. Tolerance to acid in pH 5.0-grown organisms of potentially pathogenic gram-negative bacteria. Lett Appl Microbiol. 1995; 21: 359-363. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8554762
- Urdaneta V, Casadesús J. Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med. 2017; 4: 163. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29043249
- Segata N, Haake SK, Mannon P, Lemon KP, Waldron L. et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012; 13: R42. PubMed: https://pubmed.ncbi.nlm.nih.gov/22698087/
- Choksi Y, Vaezi MF. Preliminary esophageal microbiome studies prompt important scientific questions editorial. Clin Transl Gastroenterol. 2018; 9: 135. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29807991
- Lu CL, Lang HC, Luo JC, Liu CC, Lin HC, et al. Increasing trend of the incidence of esophageal squamous cell carcinoma, but not adenocarcinoma, in Taiwan. Cancer Causes Control. 2010; 21: 269-274. PubMed: https://pubmed.ncbi.nlm.nih.gov/19866363/
- Cook MB, Chow WH, Devesa SS. Oesophageal cancer incidence in the United States by race, sex, and histologic type, 1977-2005. Br J Cancer. 2009; 101: 855-859. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19672254
- Abbas G, Krasna M. Overview of esophageal cancer. Ann Cardiothorac Surg. 2017; 6: 131-136. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28447001
- Abnet CC, Arnold M, Wei WQ. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology. 2018;154: 360-373. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28823862
- Kamangar F, Chow WH, Abnet CC, Dawsey SM. Environmental Causes of Esophageal Cancer. Gastroenterol Clin North Am. 2009;38: 27-57. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19327566
- Engel LS, Chow WH, Vaughan TL, et al. Population attributable risks of esophageal and gastric cancers. J Natl Cancer Inst. 2003;95(18): 1404-1413. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/13130116
- Susan G. Urba M. Adenocarcinoma of the Esophagus: Risk Factors and Prevention. 2000.
- Bhat S, Coleman HG, Yousef F, et al. Risk of malignant progression in Barrett’s Esophagus patients: Results from a large population-based study. J Natl Cancer Inst. 2011; 103: 1049-1057. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21680910
- Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst. 2005; 97: 142-146. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15657344
- Yang CS, Chen X, Tu S. Etiology and Prevention of Esophageal Cancer. Gastrointest Tumors. 2016; 3: 3-16. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5040887/
- Maghsudlu M, Farashahi Yazd E. Heat-induced inflammation and its role in esophageal cancer. J Dig Dis. 2017; 18: 431-444. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28749599
- Varela-Rey M, Woodhoo A, Martinez-Chantar ML, Mato JM, Lu SC. Alcohol, DNA methylation, and cancer. Alcohol Res Curr Rev. 2012; 35: 25-35. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3860423/
- Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7: 599-612. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17646865
- Li X, Li H, Dong X, Wang X, Zhu J, et al. Expression of NF-κB and TLR-4 is associated with the occurrence, progression and prognosis of esophageal squamous cell carcinoma. Int J Clin Exp Pathol. 2018; 11: 5850-5859. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6963060/
- Chen X, Winckler B, Lu M, Cheng H, Yuan Z, et al. Oral Microbiota and Risk for Esophageal Squamous Cell Carcinoma in a High-Risk Area of China. Wei Q-Y, ed. PLoS One. 2015; 10: e0143603. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4671675/
- Peters BA, Wu J, Pei Z, Yang L, Purdue MP, et al. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 2017; 77: 6777-6787. PubMed: https://pubmed.ncbi.nlm.nih.gov/29196415/
- Yu G, Gail MH, Shi J, et al. Association between upper digestive tract microbiota and cancer-predisposing states in the esophagus and stomach. Cancer Epidemiol Biomarkers Prev. 2014; 23: 735-741. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24700175
- Narikiyo M, Tanabe C, Yamada Y, et al. Frequent and preferential infection of Treponema denticola, Streptococcus mitis, and Streptococcus anginosus in esophageal cancers. Cancer Sci. 2004; 95: 569-574. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15245592
- Michaud DS, Fu Z, Shi J, Chung M. Periodontal Disease, Tooth Loss, and Cancer Risk. Epidemiol Rev. 2017; 39: 49-58. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28449041
- Tateda M, Shiga K, Saijo S, et al. Streptococcus anginosus in head and neck squamous cell carcinoma: implication in carcinogenesis. Int J Mol Med. 2000; 6: 699-703. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11078831
- Malinowski B, Wȩsierska A, Zalewska K, et al. The role of Tannerella forsythia and Porphyromonas gingivalis in pathogenesis of esophageal cancer. Infect Agent Cancer. 2019; 14: 3. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6352356/
- Lv J, Guo L, Liu JJ, Zhao HP, Zhang J, et al. Alteration of the esophageal microbiota in Barrett’s esophagus and esophageal adenocarcinoma. World J Gastroenterol. 2019; 25: 2149-2161. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31143067
- Elliott DRF, Walker AW, O’Donovan M, Parkhill J, Fitzgerald RC. A non-endoscopic device to sample the oesophageal microbiota: a case-control study. Lancet Gastroenterol Hepatol. 2017; 2: 32-42. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28404012
- Zaidi AH, Kelly LA, Kreft RE, Barlek M, Omstead AN, et al. Associations of microbiota and toll-like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer. 2016; 16: 52. PubMed: https://pubmed.ncbi.nlm.nih.gov/26841926/
- Morris CD, Armstrong GR, Bigley G, Green H, Attwood SEA. Cyclooxygenase-2 Expression in The Barrettʼs Metaplasia–Dysplasia–Adenocarcinoma Sequence. Am J Gastroenterol. 2001; 96: 990-996. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11316217
- Gallimidi AB, Fischman S, Revach B, et al. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget. 2015; 6: 22613-22623. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26158901
- Sepehr A, Kamangar F, Fahimi S, Saidi F, Abnet CC, et al. Poor oral health as a risk factor for esophageal squamous dysplasia in northeastern Iran. Anticancer Res. 2005; 25: 543-546. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15816626
- Schmidt BL, Kuczynski J, Bhattacharya A, Huey B, Corby PM, et al. Changes in abundance of oral microbiota associated with oral cancer. PLoS One. 2014; 9: e98741. PubMed: https://pubmed.ncbi.nlm.nih.gov/24887397/
- Jovanovic A, van der Tol IGH, Kostense PJ, et al. Second respiratory and upper digestive tract cancer following oral squamous cell carcinoma. Eur J Cancer Part B Oral Oncol. 1994;30: 225-229. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7950835
- Simple M, Suresh A, Das D, Kuriakose MA. Cancer stem cells and field cancerization of Oral squamous cell carcinoma. Oral Oncol. 2015; 51: 643-651. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25920765
- Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013; 105: 1907-1911. PubMed:
- Nasrollahzadeh D, Malekzadeh R, Ploner A, Shakeri R, Sotoudeh M, et al. Variations of gastric corpus microbiota are associated with early esophageal squamous cell carcinoma and squamous dysplasia. Sci Rep. 2015; 5: 8820. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24316595
- Homann N, Kärkkäinen P, Koivisto T, Nosova T, Jokelainen K, et al. Effects of acetaldehyde on cell regeneration and differentiation of the upper gastrointestinal tract mucosa. J Natl Cancer Inst. 1997; 89: 1692-1697. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9390538
- Homann N, Tillonen J, Rintamäki H, Salaspuro M, Lindqvist C, et al. Poor dental status increases acetaldehyde production from ethanol in saliva: A possible link to increased oral cancer risk among heavy drinkers. Oral Oncol. 2001; 37: 153-158. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11167142
- Matejcic M, Gunter MJ, Ferrari P. Alcohol metabolism and oesophageal cancer: a systematic review of the evidence. Carcinogenesis. 2017; 38: 859-872. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28645180
- Bull-Otterson L, Feng W, Kirpich I, et al. Metagenomic Analyses of Alcohol Induced Pathogenic Alterations in the Intestinal Microbiome and the Effect of Lactobacillus rhamnosus GG Treatment. PLoS One. 2013; 8. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23326376
- Ying W, Jing T, Bing C, Baifang W, Dai Z, et al. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol Med Rep. 2014; 9: 2352-2356. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24718485
- Lippai D, Bala S, Catalano D, Kodys K, Szabo G. Micro-RNA-155 Deficiency Prevents Alcohol-Induced Serum Endotoxin Increase and Small Bowel Inflammation in Mice. Alcohol Clin Exp Res. 2014; 38: 2217-2224. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4260268/
- Collins SM. The immunomodulation of enteric neuromuscular function: implications for motility and inflammatory disorders. Gastroenterology. 1996; 111: 1683-1699. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8942751
- Isomoto H, Nishi Y, Kanazawa Y, et al. Immune and inflammatory responses in GERD and lansoprazole. J Clin Biochem Nutr. 2007; 41: 84-91. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18193101
- Rieder F, Cheng L, Harnett KM, et al. Gastroesophageal Reflux Disease-Associated Esophagitis Induces Endogenous Cytokine Production Leading to Motor Abnormalities. Gastroenterology. 2007; 132: 154-165. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17241868
- Manetti M, Milia AF, Benelli G, Messerini L, Matucci-Cerinic M, Ibba-Manneschi L. The gastric wall in systemic sclerosis patients: A morphological study. Ital J Anat Embryol. 2010; 115: 115-121. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21073000
- Pajecki D, Zilberstein B, Cecconello I, Dos Santos MAA, Yagi OK, Gama-Rodrigues JJ. Larger amounts of nitrite and nitrate-reducing bacteria in megaesophagus of Chagas’ disease than in controls. J Gastrointest Surg. 2007; 11: 199-203. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17390173
- Pajecki D, Zilberstein B, dos Santos MAA, et al. Megaesophagus microbiota: a qualitative and quantitative analysis. J Gastrointest Surg. 6: 723-729. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12399062
- Ge X, Zhao W, DIng C, Tian H, Xu L, et al. Potential role of fecal microbiota from patients with slow transit constipation in the regulation of gastrointestinal motility. Sci Rep. 2017; 7: 441. PubMed: https://pubmed.ncbi.nlm.nih.gov/28348415/
- Cherbut C. Motor effects of short-chain fatty acids and lactate in the gastrointestinal tract. Proc Nutr Soc. 2003; 62: 95-99. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12740048
- Talotta R, Atzeni F, Ditto MC, Gerardi MC, Sarzi-Puttini P. The Microbiome in Connective Tissue Diseases and Vasculitides: An Updated Narrative Review. J Immunol Res. 2017; 2017: 6836498. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28835902
- Andréasson K, Alrawi Z, Persson A, Jönsson G, Marsal J. Intestinal dysbiosis is common in systemic sclerosis and associated with gastrointestinal and extraintestinal features of disease. Arthritis Res Ther. 2016; 18: 278. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27894337
- Volkmann ER, Hoffmann-Vold AM, Chang YL, et al. Systemic sclerosis is associated with specific alterations in gastrointestinal microbiota in two independent cohorts. BMJ Open Gastroenterol. 2017; 4: e000134. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28761687
- Patrone V, Puglisi E, Cardinali M, et al. Gut microbiota profile in systemic sclerosis patients with and without clinical evidence of gastrointestinal involvement. Sci Rep. 2017;7: 14874. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29093509
- Bellocchi C, Fernández-Ochoa Á, Montanelli G, et al. Microbial and metabolic multi-omic correlations in systemic sclerosis patients. Ann N Y Acad Sci. 2018; 1421: 97-109. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29749635
- Dellon ES, Gonsalves N, Hirano I, Furuta GT, Liacouras CA, Katzka DA. ACG clinical guideline: Evidenced based approach to the diagnosis and management of esophageal eosinophilia and eosinophilic esophagitis (EoE). Am J Gastroenterol. 2013; 108: 679-692. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23567357
- Torrijos EG, Gonzalez-Mendiola R, Alvarado M, Avila R, Prieto-Garcia A, et al. Eosinophilic esophagitis: Review and update. Front Med. 2018; 5: 247. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6192373/
- Spechler SJ. Eosinophilic esophagitis: novel concepts regarding pathogenesis and clinical manifestations. J Gastroenterol. 2019;54: 837-844. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31342146
- Simon D, Straumann A, Schoepfer AM, Simon H-U. Current concepts in eosinophilic esophagitis. Allergo J Int. 2017; 26: 258-266. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5660132/
- Sherrill JD, Kiran KC, Blanchard C, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun. 2014; 15: 361-369. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24920534
- Rieder F, Nonevski I, Ma J, Ouyang Z, West G, et al. T-helper 2 cytokines, transforming growth factor β1, and eosinophil products induce fibrogenesis and alter muscle motility in patients with eosinophilic esophagitis. Gastroenterology. 2014; 146: 1266-1277. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24486052
- Harris JK, Fang R, Wagner BD, Choe HN, Kelly CJ, et al. Esophageal microbiome in eosinophilic esophagitis. PLoS One. 2015; 10: e0128346.. PubMed: https://pubmed.ncbi.nlm.nih.gov/26020633/
- Benitez AJ, Hoffmann C, Muir AB, et al. Inflammation-associated microbiota in pediatric eosinophilic esophagitis. Microbiome. 2015; 3: 23. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4450515/
- Shin NR, Whon TW, Bae JW. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015; 33: 496-503. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26210164
- Hiremath G, Shilts MH, Boone HH, et al. The Salivary Microbiome Is Altered in Children With Eosinophilic Esophagitis and Correlates With Disease Activity. Clin Transl Gastroenterol. 2019;10: e00039. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31107724
- Kaur S, Kaur S. Bacteriocins as potential anticancer agents. Front Pharmacol. 2015; 6: 272. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4639596/
- Kumariya R, Garsa AK, Rajput YS, Sood SK, Akhtar N, et al. Bacteriocins: Classification, synthesis, mechanism of action and resistance development in food spoilage causing bacteria. Microb Pathog. 2019; 128: 171-177. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30610901
- Mathiesen G, Huehne K, Kroeckel L, Axelsson L, Eijsink VGH. Characterization of a new bacteriocin operon in sakacin P-producing Lactobacillus sakei, showing strong translational coupling between the bacteriocin and immunity genes. Appl Environ Microbiol. 2005; 71: 3565-3574. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1169027/
- Heng NCK, Tagg JR. What’s in a name? Class distinction for bacteriocins. Nat Rev Microbiol. 2006; 4: 160-160.
- Okuda KI, Zendo T, Sugimoto S, et al. Effects of bacteriocins on methicillin-resistant Staphylococcus aureus biofilm. Antimicrob Agents Chemother. 2013;57: 5572-5579. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23979748
- Corr SC, Li Y, Riedel CU, O’Toole PW, Hill C, et al. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc Natl Acad Sci U S A. 2007; 104: 7617-7621. PubMed: https://pubmed.ncbi.nlm.nih.gov/17456596/
- Joo NE, Ritchie K, Kamarajan P, Miao D, Kapila YL. Nisin, an apoptogenic bacteriocin and food preservative, attenuates HNSCC tumorigenesis via CHAC1. Cancer Med. 2012; 1: 295-305. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23342279
- Kamarajan P, Hayami T, Matte B, et al. Nisin ZP, a bacteriocin and food preservative, inhibits head and neck cancer tumorigenesis and prolongs survival. PLoS One. 2015; 10: e0131008. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26132406
- Lobo LA, Benjamim CF, Oliveira AC. The interplay between microbiota and inflammation: lessons from peritonitis and sepsis. Clin Transl Immunol. 2016; 5: e90. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27525063
- Sorini C, Cardoso RF, Gagliani N, Villablanca EJ. Commensal bacteria-specific CD4+T cell responses in health and disease. Front Immunol. 2018; 9: 2667. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30524431
- Siracusa F, Schaltenberg N, Villablanca EJ, Huber S, Gagliani N. Dietary habits and intestinal immunity: From food intake to CD4+TH cells. Front Immunol. 2019; 10: 3177. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30697217
- Selling J, Swann P, Madsen LR, Oswald J. Improvement in Gastroesophageal Reflux Symptoms From a Food-grade Maltosyl-isomaltooligosaccharide Soluble Fiber Supplement: A Case Series. Integr Med (Encinitas). 2018; 17: 40-42. PubMed:
http://www.ncbi.nlm.nih.gov/pubmed/31043918 - Gomi A, Yamaji K, Watanabe O, Yoshioka M, Miyazaki K, et al. Bifidobacterium bifidum YIT 10347 fermented milk exerts beneficial effects on gastrointestinal discomfort and symptoms in healthy adults: A double-blind, randomized, placebo-controlled study. J Dairy Sci. 2018; 101: 4830-4841. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29573807
- Waller PA, Gopal PK, Leyer GJ, et al. Dose-response effect of Bifidobacterium lactis HN019 on whole gut transit time and functional gastrointestinal symptoms in adults. Scand J Gastroenterol. 2011;46: 1057-1064. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21663486
- Nakae H, Tsuda A, Matsuoka T, Mine T, Koga Y. Gastric microbiota in the functional dyspepsia patients treated with probiotic yogurt. BMJ Open Gastroenterol. 2016; 3: e000109. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27752337
- Ohtsu T, Takagi A, Uemura N, et al. The Ameliorating Effect of Lactobacillus gasseri OLL2716 on Functional Dyspepsia in Helicobacter pylori -Uninfected Individuals: A Randomized Controlled Study. Digestion. 2017; 96: 92-102.
- Cheng J, Ouwehand AC. Gastroesophageal reflux disease and probiotics: A systematic review. Nutrients. 2020; 12: 132. PubMed: https://pubmed.ncbi.nlm.nih.gov/31906573/
- Sun QH, Wang HY, Sun SD, Zhang X, Zhang H. Beneficial effect of probiotics supplements in reflux esophagitis treated with esomeprazole: A randomized controlled trial. World J Gastroenterol. 2019; 25: 2110-2121. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31114137
- Zmora N, Zilberman-Schapira G, Suez J, et al. Personalized Gut Mucosal Colonization Resistance to Empiric Probiotics Is Associated with Unique Host and Microbiome Features. Cell. 2018;174: 1388-1405. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6993818/
- Rao SSC, Bhagatwala J. Small Intestinal Bacterial Overgrowth. Clin Transl Gastroenterol. 2019; 10: e00078. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31584459
- Young Chang J, Kim SE, Kim TH, Woo SY, Ryu MS, et al. Emergence of rifampin-resistant staphylococci after rifaximin administration in cirrhotic patients. 2017. 12: e0186120. PubMed: https://pubmed.ncbi.nlm.nih.gov/28982166/
- Mcdonald LC, Gerding DN, Johnson S, Bakken JS, Carroll KC, et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis Clin Pract Guidel Clostridium difficile Infect • CID. 2018: 66: e1-e48. PubMed: https://pubmed.ncbi.nlm.nih.gov/29462280/
- Kommineni S, Bretl DJ, Lam V, Chakraborty R, Hayward M, et al. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature. 2015; 526: 719-722. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26479034
- Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017; 357: 1156-1160. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28912244
- Fahim HA, Khairalla AS, El-Gendy AO. Nanotechnology: A valuable strategy to improve bacteriocin formulations. Front Microbiol. 2016; 7: 1385. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5026012/