
More Information
Submitted: 09 March 2020 | Approved: 20 May 2020 | Published: 21 May 2020
How to cite this article: Bajaj A. Frequent, Genetic Polyps-Familial Adenomatous Polyposis. Ann Clin Gastroenterol Hepatol. 2020; 4: 015-019.
DOI: 10.29328/journal.acgh.1001017
Copyright License: © 2020 Bajaj A, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Frequent, Genetic Polyps-Familial Adenomatous Polyposis
Anubha Bajaj*
New Delhi 110027, India
*Address for Correspondence: Dr. Anubha Bajaj, A-1, Rajouri Garden, New Delhi 110027, India, Tel: 00911141446785; Email: [email protected]
Preface
Familial adenomatous polyposis is an autosomal dominant syndrome of variable penetration and constitutes the second frequent inherited syndrome enunciating the emergence of a colorectal carcinoma. The syndrome is accompanied by exemplification of defective adenomatous polyposis coli (APC) gene located upon chromosome 5q21 with a prototypic denomination of colonic adenomatous polyps usually exceeding a > 100. Incriminated individuals develop innumerable colonic and rectal polyps, particularly during early teenage years and are accompanied by an almost 100% possible emergence of colorectal carcinoma within 40 years in untreated subjects [1]. Prophylactic colectomy is advisable to substantially reduce possible occurrence of colorectal carcinoma. Familial adenomatous polyposis is concurrent with associated neoplasms such as gastric or duodenal cancer, hepatoblastoma or desmoid tumour along with a probable emergence of extra-colonic carcinomas [1,2].
Disease characteristics
As a frequent polyposis syndrome enunciating genetic malformations on account of a germline mutation within the tumour suppressor adenomatous polyposis coli (APC) gene situated on chromosome 5q21, familial adenomatous polyposis commonly displays in excess of 100 colo-rectal adenomatous polyps. Majority of chromosomal mutations are non sense mutations although genotypic and phenotypic variation is significant on account of a magnified APC gene. Location of pertinent genetic mutation is incumbent to phenotypic variations of the syndrome as cogitated with extra-colonic disease manifestations [1,2].
Apart from a 100% lifetime possibility of emergent colorectal adenocarcinoma, familial adenomatous polyposis can be accompanied by diverse disorders such as Gardner’s syndrome, Turcot’s syndrome and attenuated familial adenomatous polyposis. Colonic, gastric (fundic gland polyp) and small intestinal polyps are a component of familial adenomatous polyposis. Additionally, occurrence of carcinoma of thyroid, gall bladder and adrenal glands can be encountered.
Familial adenomatous polyposis is additionally designated as familial polyposis coli or adenomatous polyposis coli [1,2].
Disease incidence is at an estimated 1: 7 to 1:30,000 individuals with an average incidence of 1:10000 persons. Gender equivalence is observed and males and females are equally implicated. Typically, colonic polyps emerge within the second decade with a mean age of occurrence at 15.9 years [1,2].
In the absence of an extensive, therapeutic surgical extermination, subjects comprehensively (100%) incur colonic adenocarcinoma which is discernible at an average age of 39 years. An estimated 25% candidates are diagnosed with colorectal carcinoma during disease representation [1].
Genetic assessment is necessitated in candidates delineating in excess of > ten adenomatous polyps within a singular colonoscopy. An estimated 10% to 30% individuals representing familial adenomatous polyposis are devoid of a cogent genomic mutation. Nearly 30% candidates of familial adenomatous polyposis lack a distinctive family history and manifest a de novo mutation of the APC gene [2,3]. Nevertheless, family members of subjects lacking an identifiable chromosomal mutation require an identical surveillance protocol as mandated by individuals of polyposis syndrome delineating a distinct genetic mutation.
As the syndrome is exceptional, around 1% instances of colorectal carcinoma are secondary to familial adenomatous polyposis. A milder syndromic category, the attenuated familial adenomatous polyposis (AFAP) is additionally responsible for the occurrence of colorectal carcinoma. Aforesaid syndrome is accompanied by delayed and minimal emergence of polyps and can be cogitated in adults. Subjects delineating attenuated familial polyposis syndrome demonstrate an approximate 70% possible emergence of colorectal carcinoma [2,3].
Disease pathogenesis
APC (adenomatous polyposis coli) exemplifies a tumour suppressor gene incriminated with the control of cellular cycle along with downregulation of beta catenin molecules enunciated through Wnt signalling pathway. Adenomatous polyposis coli (APC) protein is commonly implicated in apoptosis of colonic epithelial cells. Mutations of APC gene can engender an expansion of cellular component of the crypt base in addition to crypt stem cells, subsequently, clonal expansion of mutant crypt stem cells can configure adenomas and carcinomas. With the genomic mutation of APC, downregulation of beta catenin ceases with consequent stimulation of cellular growth and expansion. The two hit hypothesis delineates a germline mutation of APC gene in order to incur adenomatous polyps, especially in concurrence with inactivation of remnant, unmodified allele of the APC gene [3,4].
Autosomal dominant trait of adenomatous polyposis coli has a significant proportion of disease penetration exceeding > 90%. An estimated 20% candidates represent a de novo chromosomal mutation of APC gene accompanied by an absence of a cogent family history. Pathogenesis of colorectal adenocarcinomas within familial adenomatous polyposis is identical to the carcinogenesis cogitated within sporadically appearing colorectal adenocarcinomas, as exemplified with concurrent and discernible Kirsten rat sarcoma viral oncogene homolog (KRAS) and tumour protein (TP53) genes. Additionally, quantification of adenomatous polyps is significantly responsible for emergence of colorectal adenocarcinoma. Location of chromosomal mutation within the APC gene can influence phenotypic representation of the syndrome, specifically manifesting with quantifiable polyps, occurrence or absence of desmoid tumours along with congenital hypertrophy of retinal pigment epithelium [3,4].
Mortality in instances of non-colorectal adenocarcinomas associated with familial adenomatous polyposis is engendered on account of duodenal or ampullary carcinoma incriminating the ampulla of Vater and occurs as a consequence of a complications arising within a desmoid tumour [3].
Clinical elucidation
Clinical representation of familial adenomatous polyposis is contingent to pertinent family history. Screening is commenced with an annual endoscopy and is recommended within a younger age-group, particularly in subjects delineating a family history. Individuals demonstrating a lack of syndromic family history frequently manifest colorectal carcinoma in younger candidates or the neoplasm is discernible with screening colonoscopy. Definitive diagnosis is pertinent to the visualization of polyps exceeding > 100 on colonoscopy. Cogent extra-colonic manifestations are demonstrable with familial adenomatous polyposis [4.5].
Desmoid tumour is exceptional within the general population although an estimated 10% to 25% subjects with familial adenomatous polyposis display the neoplasm, particularly within the abdominal cavity. Individuals exhibiting a family history of desmoid tumours are associated with around 25% possible occurrence of desmoid tumour and a female predominance with a female to male proportion of 2:1. Additionally, trauma induced by abdominal surgical manoeuvers can engender a desmoid tumour. Therefore, it is advisable to defer a colectomy in young candidates delineating familial adenomatous polyposis [4,5]. Desmoid tumour is a solid, benign tumefaction arising from connective tissue. The neoplasm can enlarge to mammoth dimensions and display localized infiltration. Desmoid tumour frequently exemplifies as an enlarged, firm, painless tumefaction. Majority of tumour aggregates are spherical or elliptical and articulate an irregular tumour perimeter. As per the contemporary National Comprehensive Cancer Network (NCCN) guidelines, an annual abdominal palpation is recommended with adoption of cogent imaging modalities for candidates with a significant family history of familial adenomatous polyposis and concurrent desmoid tumours. Magnetic resonance imaging (MRI) imaging is a beneficial modality for estimating the extent of tumefaction, circumscribing anatomical structures and foci of tumour infiltration [4,5]. Ingestion of certain non steroidal anti-inflammatory agents such as sulindac and celebrex can stabilize and induce a retrogression in desmoid tumour in roughly 30% instances. Agents such as selective oestrogen receptor modulators (SERMs) can beneficially reduce desmoid tumours [4].
Gastric or duodenal polyps are enunciated in approximately 90% of candidates with familial adenomatous polyposis. In contrast to colonic polyps, gastric or duodenal polyps infrequently evolve into a frank, overt adenocarcinoma. Roughly 1% of subjects with gastric polyps enunciate gastric carcinoma. As per NCCN recommendations, endoscopy of upper gastrointestinal tract requires commencement at 20 years or 25 years. Gastric or duodenal polyps are preferentially resected with endoscopic manoeuvers, although polyps delineating a high grade dysplasia or malignant degeneration are managed with appropriate surgical eradication [5,6].
Hepatoblastoma is a neoplasm demonstrating minimal incidence in familial adenomatous polyposis. Male children beneath < five years of age are commonly implicated. Specific screening protocols are absent for determining a hepatoblastoma in familial adenomatous polyposis. Children demonstrating an enhanced possibility can be benefitted with a hepatic ultrasound and serum alpha fetoprotein levels evaluated at 3 month to 6 month interval [5,6].
Thyroid carcinoma occurs in approximately 2% subjects of familial adenomatous polyposis wherein papillary carcinoma is a frequent category. Hispanic population demonstrates an enhanced incidence of thyroid malignancies, particularly females as a majority (90%) of thyroid carcinomas arising in familial adenomatous polyposis are detected in women. Annual thyroid examination is recommended, commencing during teenage years and an annual ultra-sonographic screening can be contemplated [5,6].
Bone lesions such as osteomas, cutaneous lesions as with epidermal inclusion cysts or dental anomalies delineated with supernumerary teeth or absent teeth can be exemplified.
Gardner’s syndrome is enunciated as extra-intestinal tumours in a subset of individuals with familial adenomatous polyposis. Turcot’s syndrome delineates brain tumours, typically a medulloblastoma [5,6].
Histological elucidation
On gross examination, majority of adenomatous polyps are miniature, sessile and beneath < 0.5 cm in magnitude. Adenomatous polyps are frequent upon left gastrointestinal tract although entire segments of colon are typically incriminated. Adenomatous polyps can be flattened or depressed or depict a crater [1,2].
Adenomatous polyps are morphologically identical to classical adenoma which are categorized into varieties such as conventional, serrated, traditional serrated, flattened accompanied by conventional dysplastic alterations or mixed adenomatous polyps as configured with sessile serrated adenomatous polyp associated with conventional dysplasia. Architectural patterns are subdivided into villous adenomatous polyp wherein the villous component exceeds > 75%, tubulovillous adenomatous polyp wherein the villous component is betwixt 25% to 75% and tubular adenomatous polyps demonstrating a villous component beneath < 25% [1,2].
Proportionate villous differentiation enhances with the magnitude of adenomatous polyp. Adenomatous polyps are devoid of infiltration through superimposed muscularis mucosa into the underlying submucosa [1,2].
Hereditary colorectal cancer syndrome is a condition usually enunciated on account of defective APC gene situated upon chromosome 5q21. Subjects of familial adenomatous polyposis devoid of genomic mutation of the APC gene frequently delineate mutation of MYH gene [1,2].
Figure 1 Familial adenomatous polyp depicting crowding of mucosal glands, admixed tubular and villous articulations and few dilated crypts superimposed upon an oedematous lamina propria [10].
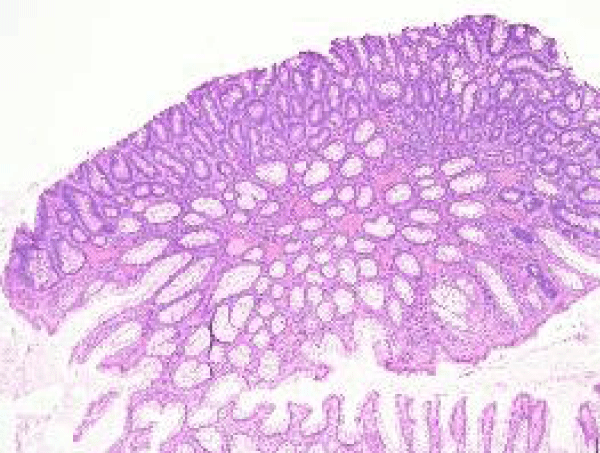
Figure 1: Courtesy: Pathology Outlines.
Figure 2 Familial adenomatous polyp delineating aggregation of tubular and villous architecture, occasional crypt dilatation and epithelial serrations [10].
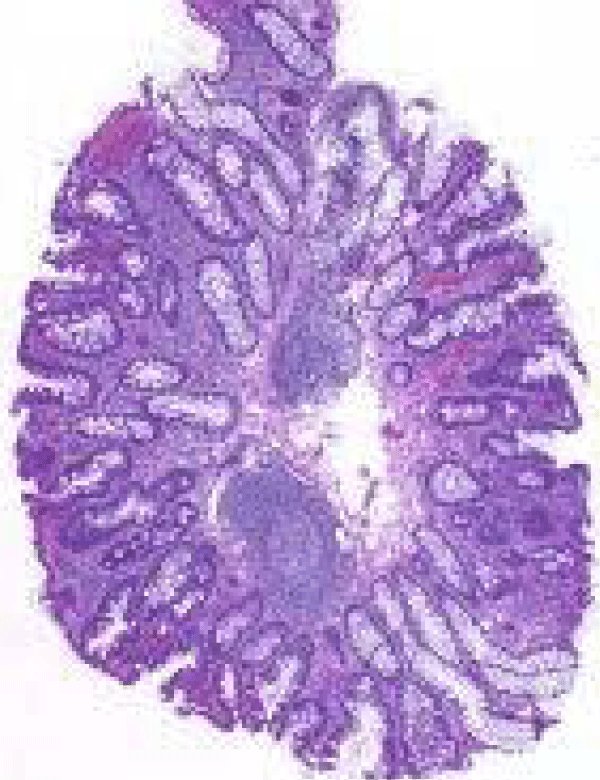
Figure 2: Courtesy: Pathology Outlines.
Figure 3 Familial adenomatous polyposis with abundant epithelial mucin, tubular arrangement, crowding of mucosal glands and absence of dysplasia [11].
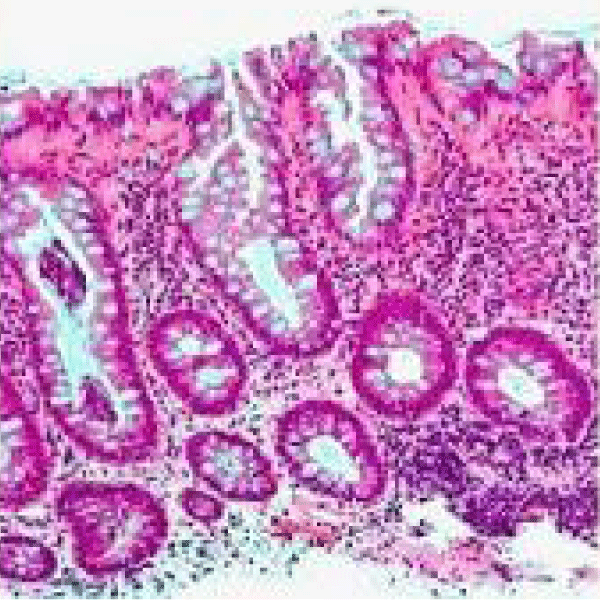
Figure 3: Semantic scholar.
Figure 4 Familial adenomatous polyposis with cystically dilated mucosal glands, admixed tubular and villous configuration and minimal epithelial mucin [12].
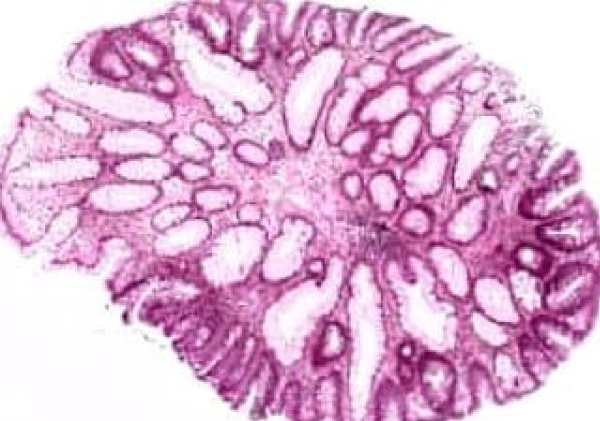
Figure 4: Courtesy: Humpath.com
Figure 5 Familial adenomatous polyposis demonstrating cystic dilatation of mucosal glands, distended chief and parietal cells and mucinous foveolar cells along with an unremarkable lamina propria [13].
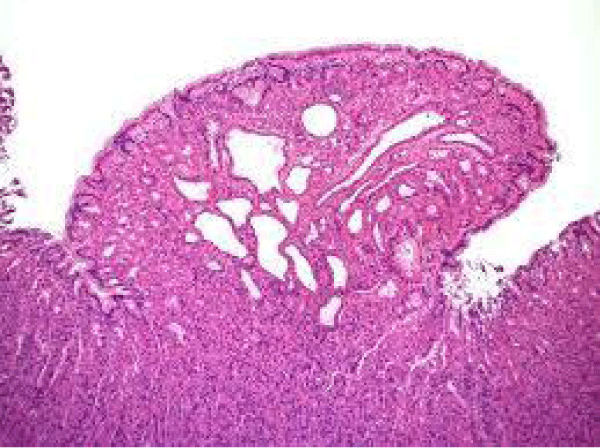
Figure 5: Courtesy: Radiopedia.com .
Figure 6 Familial adenomatous polyposis with a significant tubular and villous architecture, accumulated mucosal glands, a few of which are cystically dilated and absence of dysplasia [14].
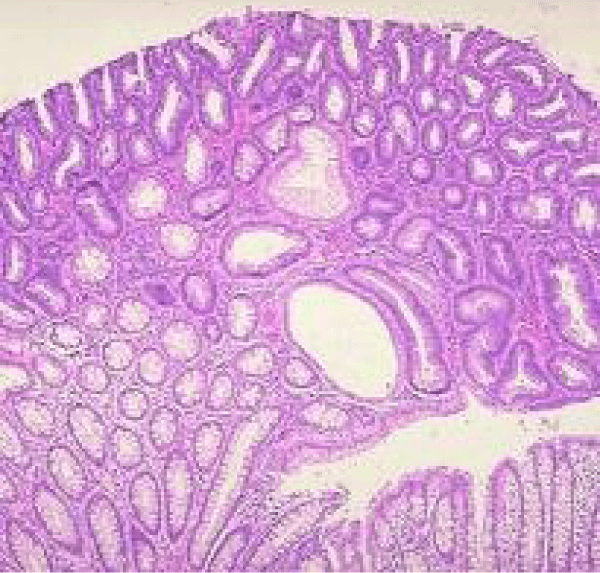
Figure 6: Courtesy: Slideplayer.com.
Figure 7 Familial adenomatous polyposis with epithelial mucin secretion, tubular arrangement and a haemorrhagic exudate in the lamina propria [15].
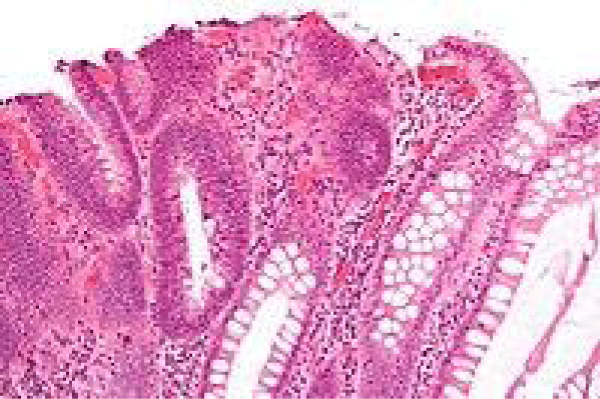
Figure 7: Courtesy: Wikipedia .
Figure 8 Familial adenomatous polyposis with a predominantly villous architecture, approximation of mucosal glands and epithelial mucin aggregates in accompaniment with absence of dysplasia [16].
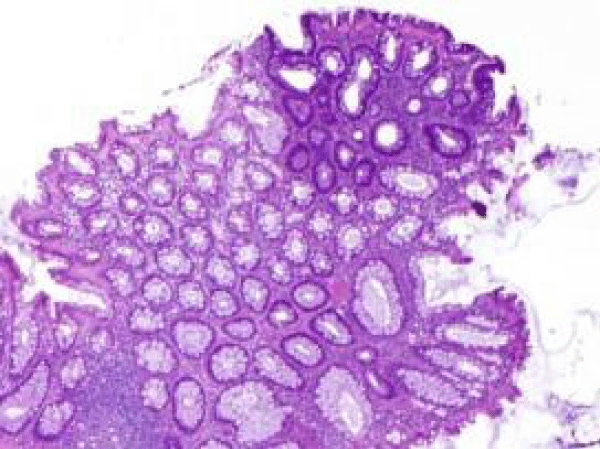
Figure 8: Courtesy: Science direct.
Figure 9 Familial adenomatous polyposis with admixture of tubular and villous configuration, cystically dilated, mucus secreting glands and an unremarkable lamina propria [16].
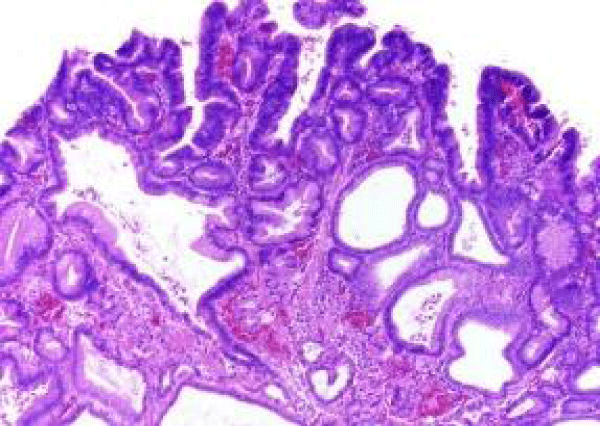
Figure 9: Courtesy: Science direct.
Figure 10 Familial adenomatous polyposis epithelial mucin accumulation, dilated oxyntic cells and parietal cells demonstrating apocrine snouts, cystic dilatation of mucosal glands and extravasation of red cells in the lamina propria [17].
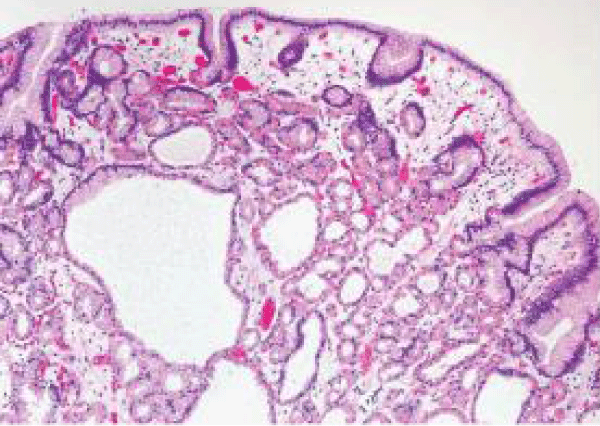
Figure 10: Courtesy: Abdominal key.
Diagnostic criterion
Cogent criterion for discerning familial adenomatous polyposis are
•occurrence of colorectal adenomatous polyps exceeding a 100 in quantification.
•or a germline mutation of the APC gene.
•or a family history of familial adenomatous and concurrent colorectal adenomas appearing below < 30 years.
•or a family history of familial adenomatous polyposis in association with minimally a singular tumefaction such as an epidermoid cyst, osteoma or desmoid tumour [5,6].
Specifically, subjects demonstrating a history of colorectal adenomas exceeding > 10, a family history of adenomatous polyposis syndrome besides a history of adenomas in concurrence with extra-colonic lesions of the familial adenomatous polyposis category mandate an assessment for adenomatous polyposis syndrome [6].
Differential diagnosis
Familial polyposis syndrome requires a segregation from attenuated familial adenomatous polyposis syndrome, a condition additionally engendered by a chromosomal mutation of the APC gene although implicated subjects demonstrate quantifiable colorectal polyps usually beneath < a 100 [1,2].
Also, differentiation from a MYH associated polyposis or a polyposis syndrome, commonly initiated by genomic mutation of MYH gene is necessitated [1].
Investigative assay
Candidates with established familial adenomatous polyposis or a strong family history of polyposis mandate an annual evaluation with endoscopy with the employment of a flexible sigmoidoscopy or colonoscopy which can be commenced at 10 years to 12 years.
Surveillance is continued until extraction of polyp is manageable with endoscopic extermination. Genetic assay is unnecessary at a preliminary age on account of a psychological detriment accompanying the genomic mutations. Upon clinical confirmation of familial adenomatous polyposis, genetic assessment is advantageous and recommended [5,6]
Therapeutic options
Familial adenomatous polyposis and attenuated familial adenomatous polyposis syndrome are preferentially managed with a comprehensive surgical resection in order to significantly reduce probable occurrence of colorectal carcinoma.
Surgical management is preferred and recommended, definitive surgical procedures are denominated by a colectomy in combination with or absence of proctectomy. Acceptable surgical manoeuvers are comprised of subtotal colectomy with ileo-rectal anastomosis, subtotal colectomy with ileostomy or total procto-colectomy with the construction of an ileo-anal pouch [6,7]. Subtotal colectomy is technically simple and minimally challenging although continued surveillance of the rectum is required. Incriminated rectal mucosa demonstrates an increased incidence of possible adenocarcinoma. Instances delineating an intact rectum necessitate an endoscopic surveillance within six months as an estimated proportion of 29% is encountered for the occurrence of rectal carcinoma within 50 years. Surgical techniques sparing the rectum can be employed for individuals displaying minimal quantification of predominantly rectal adenomatous polyps, an absence of advanced stage of rectal malignancies and an absence of discernible colorectal carcinoma whilst undertaking a surgical resection [6,7].
Total procto-colectomy can be deemed unnecessary in appropriately screened subjects although the proportion of total procto-colectomy is around 35% in inadequately selected candidates. Total procto-colectomy as a surgical manoeuver mandates the eradication of colon and rectum along with the configuration of either an ileostomy or an ileo-anal pouch. An absence of surveillance is acceptable with aforesaid manoeuvers. Total procto-colectomy with reconstruction of ileo-anal pouch can elevate proportionate infertility and urinary dysfunction. Although urinary incontinence is not observed, enhanced stool urgency can be discerned with an articulated ileo-anal pouch [8,9].
Prophylactic colectomy is recommended within the late second or early third decade. Monitoring of remnants of rectal stump is advisable. Candidates with familial adenomatous polyposis (FAP) subjected to a consistent screening of the colon and rectum followed by a surgical resection demonstrate a significantly decimated proportion of individual mortality on account of emerging colorectal malignancies. Children displaying a possible occurrence of familial adenomatous polyposis require a sigmoidoscopic evaluation annually or once in every two years, usually commencing at 10 years to 12 years. Family and blood relatives mandate a screening [8,9].
Non-surgical therapeutic modalities are adopted in order to defer application of surgical procedures, although the outcomes are limited. Non-steroidal anti-inflammatory drugs such as sulindac can reduce the quantity of adenomatous polyps by approximately 50% and magnitude of the polyps by 65%. Discontinuation of the agent is followed by reoccurrence of the polyps. Candidates with retained rectum can be administered the nonsteroidal agent in order to decimate quantifiable polyps within the rectum [8,9]. Amplified doses of selective COX-2 inhibitor celecoxib can be administered and are accompanied by an estimated 30% decline in quantifiable polyps. Medical therapy can assist in disease stabilization.
Annual surveillance remains essential in subjects delineating a retained rectum. Preliminary endoscopic surveillance is critical for confirming an appropriately timed surgical resection [8,9].
Administration of non-steroidal anti-inflammatory drugs (NSAID) and aspirin influences and decimates reoccurrence of polyps following subtotal colectomy [8].
Emergence of extra- colonic manifestations can be discerned with intensive screening and closely monitored clinical follow up. With the adoption of recommended surveillance protocols and adequate surgical resection of familial adenomatous polyposis, a substantial reduction of possible occurrence of colorectal carcinoma and adjunctive malignancies can be achieved [8,9].
- Kanth P, Grimmett J, Champine M, Burt R, Samadder NJ. Hereditary Colorectal Polyposis and Cancer Syndromes: A Primer on Diagnosis and Management. Am J Gastroenterol. 2017; 112:1509-1525. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28786406
- Chintalacheruvu LM, Shaw T, Buddam A, Diab O, Kassim T, et al. Major hereditary gastrointestinal cancer syndromes: a narrative review. J Gastrointestinal Liver Dis. 2017; 26: 157-163. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28617886
- Carr S, Kasi A. Familial adenomatous polyposis. StatPearls Publishing 2020. PubMed: https://www.ncbi.nlm.nih.gov/books/NBK538233/
- Sammour T, Hayes IP, Hill AG, Macrae FA, Winter DC. Familial colorectal cancer syndrome: an overview of clinical management. Expert Rev Gastroenterol Hepatol. 2015; 9: 757-764. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25779305
- DE Marchis ML, Tonelli F, Quaresmini D, Lovero D, Della-Morte D, et al. Desmoid tumours in familial adenomatous polyposis. Anticancer Res. 2017; 37: 3357-3366. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28668823
- Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, et al. American College of Gastroenterology ACG: clinical guidelines: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015; 110: 223-262. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25645574
- Chittleborough TJ, Warrier SK, Heriot AG, Kalady M, Church J. Dispelling misconceptions in the management of familial adenomatous polyposis. ANZ J Surg. 2017; 87: 441-445. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/28266097
- Waller A, Findeis S, Lee MJ. Familial adenomatous polyposis. J Paediatr Genet. 2016; 5: 78-83. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27617147
- Dinarvand P, Davaro EP, Doan JV, Ising ME, Evans NR, et al. Familial adenomatous polyposis syndrome: an update and review of extra-intestinal manifestations. Arch Pathol Lab Med. 2019; 143: 1382-1398. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31070935